L’accès précoce aux médicaments innovants, nouveau dispositif mis en place en juillet 2021, répond à une réglementation précise. Il repose sur des décisions conjointes de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) et de la Haute Autorité de santé (HAS) et implique des réflexions éthiques et sociétales. Illustration par quelques situations dans le domaine de l’oncohématologie.
L’accès précoce consiste en l’utilisation d’un médicament dans une indication thérapeutique précise qui n’est pas encore couverte par une autorisation de mise sur le marché (AMM), à titre exceptionnel et en dehors du cadre d’un essai clinique.
Selon l’article L5121-12 du code de la santé publique et selon la HAS, cinq conditions doivent être remplies pour accorder un accès précoce à un médicament donné :1, 2
– la maladie doit être « rare, grave ou invalidante ». La gravité et le caractère invalidant sont évalués au regard des symptômes, de l’atteinte des organes, du taux de mortalité et de l’impact sur la qualité de vie ;
– « la mise en œuvre du traitement ne peut pas être différée » ;
– « il n’existe pas de traitement approprié », c’est-à-dire une alternative thérapeutique répondant à quatre critères (recommandée au même niveau de la stratégie thérapeutique, accessible en pratique courante, prise en charge par la solidarité nationale, disposant de données d’efficacité et de tolérance satisfaisantes ne suggérant pas de perte de chance par rapport au médicament faisant l’objet de l’accès précoce) ;
– le médicament est « présumé innovant, notamment au regard d’un éventuel comparateur cliniquement pertinent ». Un médicament est présumé innovant s’il répond à trois critères (apport d’un changement substantiel à la prise en charge en matière d’efficacité, de qualité de vie, de tolérance, de praticité, de commodité d’emploi ou de parcours de soins ; existence d’un plan de développement adapté et de résultats cliniques étayant la présomption d’un bénéfice ; réponse à un besoin médical non ou insuffisamment couvert) ;
– « l’efficacité et la sécurité du médicament sont fortement présumées au vu des résultats d’essais thérapeutiques ».
Accélérer l’accès aux médicaments innovants
Le cancer est l’un des plus grands problèmes de santé publique dans le monde. Pour lutter contre ce fléau, améliorer le développement des médicaments anticancéreux et l’efficacité des systèmes de santé est une priorité absolue. L’accès à ces traitements, en particulier aux médicaments innovants, varie dans le monde et dépend de la puissance économique du pays et des choix politiques fondés sur les preuves scientifiques et le rapport coût-efficacité. Dans l’Union européenne (UE), cet accès passe par des décisions d’AMM centralisées prises par le comité des médicaments à usage humain de l’Agence européenne des médicaments (EMA), avant que chaque pays ne décide du remboursement en fonction de son système national de santé.
Afin d’assurer la disponibilité la plus rapide possible des innovations thérapeutiques, plusieurs systèmes ont été mis au point par la Food and Drug Administration (FDA) [fast track, priority review, breakthrough therapy] et l’EMA (priority medicines [PRIME], AMM conditionnelle). Malheureusement, étant donné la complexité et le temps que prennent ces processus réglementaires, l’accès rapide aux médicaments anticancéreux reste un défi.3, 4
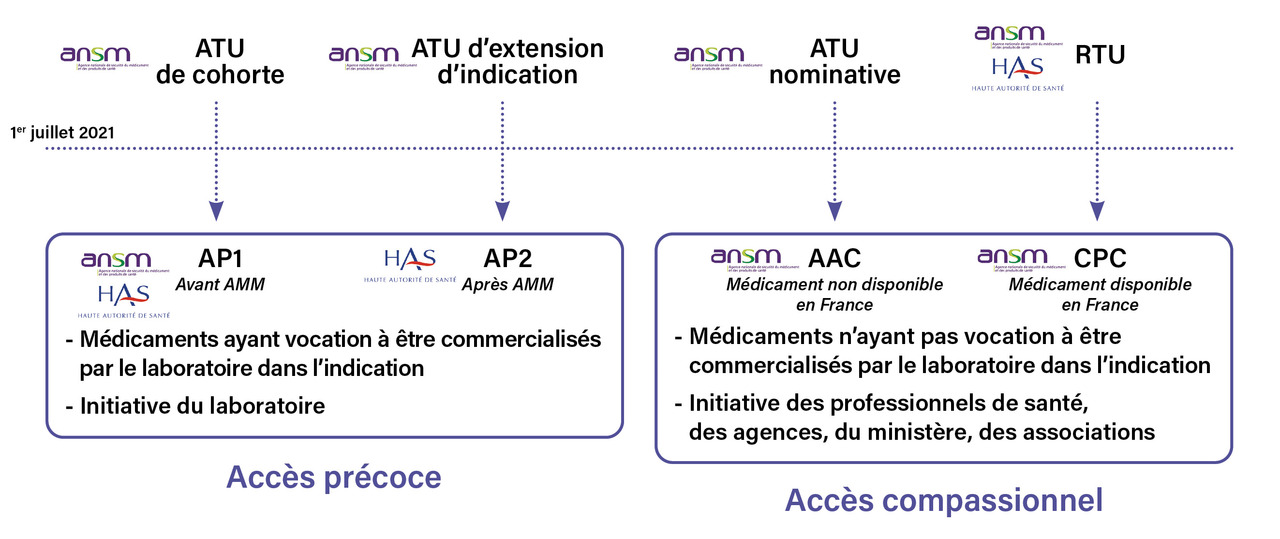
De 1994 à 2021, un système d’autorisation temporaire d’utilisation
La France a mis en place, en 1994, le système d’autorisation temporaire d’utilisation (ATU), qui permet l’utilisation exceptionnelle, après une évaluation approfondie, de traitements innovants avant l’AMM, avec une prise en charge par la solidarité nationale. On distingue trois types d’ATU :
– l’ATU nominative (ATUn), délivrée pour un patient désigné, à l’initiative du médecin prescripteur et sous sa responsabilité, pour une spécialité ne disposant pas d’une AMM ;
– l’ATU de cohorte (ATUc), délivrée pour une population de patients désignée, à la demande de l’industriel, pour une spécialité ne disposant pas d’AMM ;
– l’ATU d’extension (ATUc EI), permettant d’accorder une ATU de cohorte à un médicament qui a déjà une AMM dans une autre indication.
En complément du système d’ATU, le dispositif de recommandation temporaire d’utilisation (RTU) permet d’encadrer la prescription de médicaments dans une indication ou des conditions d’utilisation différentes de leur AMM, dans le cas où l’industriel n’a pas d’intention de déposer une demande d’AMM dans cette indication.
L’accès précoce ou l’accès compassionnel depuis juillet 2021
Depuis le 1er juillet 2021, à la suite de la loi de financement de la sécurité sociale (LFSS), ce dispositif a été fortement remanié en vue de simplifier l’accès précoce. Les objectifs étaient de renforcer l’accès aux nouvelles molécules présumées efficaces dans des maladies rares ou graves, de proposer un accès aux médicaments disposant d’une AMM dans d’autres pays et qui ne seraient pas disponibles en France, d’encadrer la prescription hors AMM afin de la sécuriser, et enfin de renforcer le recueil des données en vie réelle.
Cette réforme associe l’ANSM et la HAS dans la prise de décision afin que l’accès précoce intègre la dimension de la prise en charge par la solidarité nationale, évitant ainsi au mieux une rupture d’accès. En effet, de nombreuses thérapies innovantes ayant bénéficié d’une ATU puis approuvées par l’EMA n’obtenaient pas le remboursement en France après décision de la HAS et n’étaient donc plus disponibles. Cela créait une inégalité au sein du territoire français car certains hôpitaux prenaient la décision de prendre en charge ces thérapies en fonction de leurs décisions budgétaires et d’autres non.
À présent, il existe quatre dispositifs (
– l’accès précoce pré-AMM (AP1), qui remplace l’ATUc ;
– l’accès précoce post-AMM (AP2), qui remplace l’ATUc d’extension ;
– l’autorisation d’accès compassionnel (AAC), qui remplace l’ATUn ;
– le cadre de prescription compassionnelle (CPC), qui remplace la recommandation temporaire d’utilisation (RTU).
Une évaluation de ces accès précoces sera néanmoins nécessaire afin de déterminer si leurs performances sont aussi satisfaisantes que celles de l’ancien système. Enfin, les professionnels de santé doivent s’emparer de ces nouvelles dispositions pour augmenter le niveau de connaissances des traitements bénéficiant de l’accès précoce par un recueil le plus exhaustif possible des données de tolérance, d’efficacité et de qualité de vie, afin d’enrichir les dossiers d’enregistrement de ces médicaments.
Un système d’accès précoce performant
En France, le délai moyen entre l’AMM et l’accès des patients aux médicaments remboursables est de 530 jours,5 ce qui a donné lieu à de nombreuses critiques sur les retards d’accès à l’innovation thérapeutique par rapport à d’autres pays européens ou mondiaux comme les États-Unis. Cependant, ces critiques ne prennent pas en compte les systèmes d’accès précoce, qui permettent l’accès remboursé au médicament, avant AMM.
Entre le 1er janvier 2007 et le 31 décembre 2019, 36 médicaments sur les 64 ayant reçu une AMM en oncologie solide se sont vu attribuer une ATU, au bénéfice de 16 927 patients ; 25 médicaments ont été mis à disposition de manière précoce, 203 jours en moyenne avant l’AMM américaine et 428 jours en moyenne avant l’AMM européenne.6 En outre, une évaluation du bénéfice thérapeutique ajouté de ces médicaments a été réalisée, à l’aide de l’American Society of Clinical Oncology Value Framework (ASCO VF), de l’European Society for Medical Oncology-Magnitude Clinical Benefit Scale (ESMO-MCBS) et de l’amélioration du service médical rendu (ASMR) par la HAS. Sur les 35 médicaments évalués, 100 % ont obtenu une AMM et ont été remboursés par la solidarité nationale, et la majorité avaient un bénéfice thérapeutique ajouté d’après l’ESMO-MCBS (63 %), l’ASCO VF (57,6 %) et l’ASMR (71,8 %).7
Ainsi, non seulement le dispositif d’ATU a permis d’accéder à des médicaments innovants bien en amont des AMM européenne et américaine, mais ces médicaments ont confirmé par la suite un bénéfice supplémentaire dans la stratégie thérapeutique. Les comparaisons sont néanmoins à pondérer, en particulier par rapport aux États-Unis, qui ne régulent pas le remboursement et laissent le marché assurantiel privé effectuer cette régulation.
Des impacts sociétaux et éthiques à prendre en compte
L’accès à l’innovation doit être aussi appréhendé sous l’angle de la conservation des droits et du bien-être de la personne humaine dans une similitude avec l’implication des comités de protection des personnes en recherche clinique vis-à-vis des enjeux éthiques.8 En effet, ce qui semble judicieux ou pertinent à un industriel ou à un professionnel de santé peut l’être beaucoup moins selon le point de vue d’un patient. L’exemple des biopsies tumorales demandées de façon itérative dans un but de recherche biologique cognitive mais sans aucun intérêt particulier pour le patient en tant qu’individu unique et à risque de morbidité peut interroger, ceci d’autant plus que l’on s’adresse en oncologie à des patients avec une survie limitée au cours de laquelle la qualité de vie devrait largement primer.9
Une définition de l’innovation parfois en décalage avec l’intérêt du patient
En situation de cancers métastatiques en « énième » ligne, la majorité des patients sont en recherche d’alternatives thérapeutiques ; l’accès à une innovation peut apparaître comme un objectif pour ces malades. Mais nouveau médicament ne rime pas toujours avec gain thérapeutique en matière de survie ou de qualité de vie. Ainsi, alors que les CAR-T cells dans le myélome réfractaire peuvent apparaître comme une réelle avancée,10 proposer une thérapie guidée par un événement moléculaire à un patient en situation de maladie métastatique réfractaire n’a que peu d’intérêt à l’échelle individuelle ; cette thérapie ciblée risque en effet de dégrader la qualité de vie sans aucun bénéfice de survie et pour une réduction tumorale très incertaine et anecdotique de 0,9 % (réponse objective) pour les 6 % de patients qui ont pu recevoir une molécule ciblée.11 Le problème n’est pas la pertinence de l’approche de recherche mais l’incertitude sur le fait que le patient soit vraiment informé de l’absence de réel bénéfice attendu. Le dogme statuant que l’innovation thérapeutique ou les essais thérapeutiques innovants impliquent forcément un bénéfice pour le patient doit laisser sa place à une information juste et honnête sur les risques et bénéfices réellement attendus.
Depuis de nombreuses années, la valorisation de la recherche clinique et/ou fondamentale pour les carrières professionnelles ou les subventions se fait par l’analyse de la bibliométrie (Système d’interrogation, de gestion et d’analyse des publications scientifiques [SIGAPS]), sous-tendue par l’« impact factor » des publications. Il s’agit d’un cercle qui n’est pas toujours vertueux, impliquant des collaborations médicales et industrielles dans une stratégie assumée de communication en santé : développement d’un produit par une compagnie pharmaceutique, passage à l’essai clinique sous l’égide d’un « key opinion leader » (KOL) parfois guidé et accompagné dans ses choix par le propriétaire de la molécule, analyse des résultats par l’industriel, présentation de ces derniers en session plénière d’un grand congrès international puis publication libérée le jour de la présentation dans des journaux scientifiques majeurs. Ce système convient aux différents acteurs qui assurent la promotion d’une avancée thérapeutique (industriels et KOL) et il permet aux patients d’en bénéficier. Néanmoins, il est parfois dévoyé ou exagéré dans l’analyse du bénéfice. L’exemple récent d’une avancée considérée comme majeure dans le traitement du lymphome peut laisser dubitatif : intégration du polatuzumab vedotin dans le standard de « first-line » (1L) R-CHOP (rituximab, cyclophosphamide, hydroxydoxorubicine-vincristine, prednisone). En effet, cette avancée a bénéficié de nombreux relais dans la presse médicale spécialisée et la presse généraliste, puis d’une diffusion de l’information du KOL coordinateur vers les KOL régionaux. Cependant, l’analyse circonstanciée du réel bénéfice peut laisser perplexe : pas d’avantage en survie globale de la nouvelle procédure, un très faible avantage en survie sans progression à deux ans, une tolérance inchangée et un suivi très court qui ne permet pas, à ce jour, de considérer cette nouvelle procédure comme une réelle avancée !12
Des évaluations et des décisions réglementaires parfois éloignées de la réalité de terrain
En France, l’accès précoce est régulé administrativement par deux agences : l’ANSM et la HAS. Leur périmètre d’action diffère en fonction du type d’accès précoce (pré-AMM [AP1] ou post-AMM [AP2]), mais globalement la décision a été transférée de l’ANSM vers la HAS. L’ANSM prend en charge l’analyse du rapport bénéfices-risques alors que la HAS analyse un rapport bénéfices-risques relatif (nouvelle stratégie thérapeutique par rapport aux stratégies existantes). Cette dichotomie dans les périmètres des agences ne favorise pas la nécessaire vue globale d’un dossier.
Un exemple de ces difficultés et de l’opposition qui peut exister entre cliniciens et méthodologistes est donné par l’analyse du dossier Kimozo (témozolomide) dans les indications « en monothérapie ou en association à un inhibiteur spécifique de l’ADN topo-isomérase I (irinotécan ou topotécan) dans le traitement des patients pédiatriques âgés de 1 à 6 ans et chez les patients âgés de plus de 6 ans dans l’incapacité d’avaler le témozolomide sous forme de gélules et atteints d’un neuroblastome ». La spécialité Kimozo est une suspension buvable développée pour permettre l’administration en pédiatrie, car la spécialité Temodal (témozolomide) sous forme de gélules ne peut pas être utilisée chez le jeune enfant. L’ANSM a donc favorisé cet accès précoce pour permettre le traitement des enfants. En revanche, la commission de la Transparence (CT) a considéré que bien que la suspension buvable permette de traiter des enfants dans l’incapacité d’avaler des gélules, la mise en œuvre du traitement par Kimozo pouvait être « différée jusqu’à l’obtention des données permettant de lever les incertitudes majeures actuelles sur ce produit » et le « médicament n’est pas présumé innovant compte tenu des inconnues majeures liées à la transposabilité des données chez l’enfant et de l’absence de données de tolérance issues des essais cliniques ». Ainsi, la CT a émis un avis défavorable à la seule possibilité d’administration d’un médicament pour traiter un cancer pédiatrique sur des arguments méthodologiques en complet décalage avec la réalité clinique du terrain. Heureusement, le collège de la HAS n’a pas suivi l’avis de la CT et, in fine, un avis favorable a été donné.13
Un article, publié dans le Bulletin du Cancer, décrit l’ensemble des processus décisionnels ayant conduit à la non-publication des arrêtés de prise en charge de trois RTU en thérapie adjuvante du mélanome (nivolumab, pembrolizumab et l’association dabrafénib-tramétinib) et la perte de chance induite par cette décision. Ces traitements ont démontré des réductions du risque de récidive de 35 %, 43 % et 55 % pour des populations cibles de 2 200, 1 900 et 650 patients par an. Malgré un avis favorable au remboursement des scientifiques, des sociétés savantes et des agences sanitaires, les arrêtés de prise en charge n’ont jamais été publiés, interdisant l’utilisation de ces produits en amont de l’AMM, et privant les patients d’une potentielle guérison.14 Dans ce domaine, il est possible de considérer que les médecins sont contraints d’assumer sur le terrain des décisions de non-prise en charge pour des innovations qu’ils considèrent comme pertinentes.
La « liste en sus », qui correspond au financement des médicaments en dehors du forfait prévu par la solidarité nationale, en est aussi une illustration.
Indépendance nécessaire entre les industries de santé et les associations de patients
L’innovation thérapeutique est souvent considérée par les patients comme une avancée, la non-accessibilité pouvant être ressentie comme une perte de chance. Ces innovations bénéficient ainsi d’une communication active de la part des associations de patients. Cependant, l’une des problématiques de ces organisations est leur dépendance vis-à-vis de l’industrie pharmaceutique qui, bien souvent, les subventionne. En effet, d’après un rapport de la HAS, les industriels de santé ont déclaré avoir versé 5,8 millions d’euros à 356 associations en France en 2011.15 Dans l’analyse d’une procédure d’accès à l’innovation, il est primordial qu’une parfaite indépendance soit établie entre industriels et patients ou prescripteurs afin que l’intérêt de l’entreprise qui propose une nouveauté ne soit pas confondu avec celui du patient.
Absence de lien entre le prix d’un traitement et son bénéfice thérapeutique
Contrairement à ce qui pourrait être une évidence, le prix d’un médicament ou d’une innovation thérapeutique n’est pas lié à son efficacité ou à un service rendu. Différentes études ont clairement montré en oncologie qu’il n’y avait pas de corrélation entre le prix d’un produit de santé et les données d’évaluation comme la survie globale ou la survie sans progression.7, 16 Ce qui semble réguler le prix d’un médicament est in fine la somme que le payeur est prêt à débourser ou encore la soutenabilité ou l’acceptabilité de notre solidarité nationale en vue de la prise en charge d’un médicament.17 À titre d’exemple, on peut citer le prix des CAR-T cells, proche des 350 000 euros pour une injection, ce montant ne relevant pas du coût de fabrication ou de développement mais plutôt d’une approximation de ce que la société est prête à débourser. Les médicaments les plus chers du marché sont habituellement les produits utilisés dans des maladies orphelines ou génétiques qui concernent peu de personnes (exemple de Zolgensma [onasemnogène abéparvovec] pour le traitement d’enfants atteints d’amyotrophie spinale, vendu 2 millions d’euros environ par injection).18
L’accès précoce constitue parfois un frein à la recherche clinique
De façon étonnante, l’accès à l’innovation peut être un frein à la recherche clinique et à l’inclusion dans des essais cliniques. En effet, si un produit devient accessible en accès précoce et que, dans le même temps, une étude comparative est toujours en cours pour établir sa supériorité, il peut être délétère ou non souhaité d’inclure un patient dans l’essai comparatif, au risque de soumettre le patient au traitement comparateur semblant moins efficace. Dans une réflexion similaire, l’évaluation des essais cliniques effectuée par l’ANSM se concentre sur la notion de perte de chance à être inclus dans un essai. Ainsi, l’évolution des traitements et en particulier la possibilité d’accéder à une innovation thérapeutique peuvent rendre le bras standard d’un essai thérapeutique caduc ou délétère. Ceci a été décrit par la Ligue contre le cancer dans un avis du comité d’éthique daté du 17 janvier 2022.19
1. Legifrance. Code de la santé publique, article L5121-12. 1er juillet 2021. Disponible sur : www.legifrance.gouv.fr/codes/article_lc/LEGIARTI000041721215/
2. Haute Autorité de santé. Accès précoce à un médicament. 1er juillet 2021. Disponible sur : www.has-sante.fr/jcms/r_1500918/fr/acces-precoce-a-un-medicament
3. Johnson JR, Ning YM, Farrell A, Justice R, Keegan P, Pazdur R. Accelerated approval of oncology products: the food and drug administration experience. J Natl Cancer Inst 2011;103(8):636-44.
4. European Medicines Agency. Support for early access. Disponible sur : www.ema.europa.eu/en/human-regulatory/overview/support-early-access
5. Assemblée nationale. Proposition de loi visant à garantir à chacun un droit d’accès aux médicaments et dispositifs innovants. 17 juillet 2019. Disponible sur : www.assemblee-nationale.fr/dyn/15/textes/l15b2153_proposition-loi#
6. Jacquet E, Kerouani-Lafaye G, Grude F, Goncalves S, Lorence A, Turcry F, et al. Comparative study on anticancer drug access times between FDA, EMA and the French temporary authorisation for use program over 13 years. Eur J Cancer 2021;149:82-90.
7. Pham FY, Jacquet E, Taleb A, Monard A, Kerouani-Lafaye G, Turcry F, et al. Survival, cost and added therapeutic benefit of drugs granted early access through the French temporary authorization for use program in solid tumors from 2009 to 2019. Int J Cancer 2022 May 23. doi: 10.1002/ijc.34129. Epub ahead of print. PMID: 35603979.
8. David J Rothman. Strangers at the bedside: A history of how law and bioethics transformed medical decision making. 1991.
9. Overman MJ, Modak J, Kopetz S, Murthy R, Yao JC, Hicks ME, et al. Use of research biopsies in clinical trials: are risks and benefits adequately discussed? J Clin Oncol 2013;31(1):17-22.
10. Haute Autorité de santé. ABECMA. Décision d’accès précoce. 3 décembre 2021. Disponible sur : www.has-sante.fr/jcms/p_3303102/fr/abecma-idecabtagene-vicleucel-myelome-multiple
11. Trédan O, Wang Q, Pissaloux D, Cassier P, de la Fouchardière A, Fayette J, et al. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: analysis from the ProfiLER trial. Ann Oncol 2019; 30(5):757-65.
12. Tilly H, Morschhauser F, Sehn LH, Friedberg JW, Trněný M, Sharman JP, et al. Polatuzumab vedotin in previously untreated diffuse large B-cell lymphoma. N Engl J Med 2022;386(4):351-63.
13. Haute Autorité de santé. Kimozo. Décision d’accès précoce. 12 avril 2022. Disponible sur : www.has-sante.fr/jcms/p_3329593/fr/kimozo-temozolomide
14. Auger C, Guillot B, Monard A, Albin N. Délais administratifs des RTU, effet sur l’accès à l’innovation dans le mélanome, Bulletin du Cancer 2022;109(1):28-37.
15. Haute Autorité de santé. Publication des aides versées par les industries de santé aux associations de patients. Rapport 2012 portant sur les aides versées au titre de l’année civile 2011. 12 octobre 2012. Disponible sur : www.has-sante.fr/upload/docs/application/pdf/2012-10/note_financement_industries_assos_chiffres_2011_vdef.pdf
16. Rodwin MA, Mancini J, Duran S, Jalbert AC, Viens P, Maraninchi D, et al. The use of added benefit’ to determine the price of new anti-cancer drugs in France, 2004-2017. Eur J Cancer 2021;145:11‑8.
17. Ministère des Affaires sociales et de la Santé. Accès universel aux traitements innovants contre l’hépatite C. Après avoir permis l’accès de tous les malades aux traitements, Marisol Touraine obtient une baisse de prix importante. 31 mars 2017. Disponible sur : solidaritessante.gouv.fr/IMG/pdf/17_03_31__cp_concretisation_acces_universel_hepatite_c.pdf
18. Prescrire. Zolgensma : le médicament de tous les excès. 1er décembre 2019. Disponible sur : www.prescrire.org/fr/3/31/58118/0/NewsDetails.aspx
19. Comité Éthique et Cancer. Essais cliniques en cancérologie : garantir la validité des traitements utilisés comme comparateurs. 17 janvier 2022. Disponible sur : www.ethique-cancer.fr/avis/avis-ndeg40
Dans cet article
- Accélérer l’accès aux médicaments innovants
- Un système d’accès précoce performant
- Des impacts sociétaux et éthiques à prendre en compte
- Une définition de l’innovation parfois en décalage avec l’intérêt du patient
- Des évaluations et des décisions réglementaires parfois éloignées de la réalité de terrain
- Indépendance nécessaire entre les industries de santé et les associations de patients
- Absence de lien entre le prix d’un traitement et son bénéfice thérapeutique
- L’accès précoce constitue parfois un frein à la recherche clinique