L’hypophyse est composée de deux lobes :
- l’antéhypophyse sécrète l’hormone de croissance (GH), les gonadotrophines (hormone lutéinisante [LH] et hormone folliculostimulante [FSH]), la thyréostimuline (TSH), la prolactine et l’hormone adrénocorticotrope (ACTH). La sécrétion de ces hormones est stimulée par les hormones hypothalamiques : growth hormone-releasing hormone, ou hormone de libération de l’hormone de croissance (GHRH) pour la GH, hormone thyréotrope (TRH) pour la TSH, gonadotropin-releasing hormone, ou gonadolibérine (GnRH) pour la FSH et la LH. La sécrétion de prolactine est inhibée en continu par la dopamine ;
- la posthypophyse est le siège de la sécrétion d’hormone antidiurétique (ADH) et d’ocytocine.
- un syndrome tumoral en cas de macro-adénome (céphalées, troubles visuels) ;
- un syndrome d’hypersécrétion hormonale (hyperprolactinémie, acromégalie, hypercorticisme, exceptionnellement hyperthyroïdie) ;
- un syndrome d’insuffisance antéhypophysaire.
Épidémiologie
C’est l’adénome à prolactine qui est le plus fréquent (57 %), suivi des adénomes non fonctionnels dans 11 % des cas et de l’acromégalie dans 11 % des cas. L’adénome corticotrope représente 2 % des cas.
Les adénomes non fonctionnels sont plus fréquents chez les hommes (57 %), et les prolactinomes chez les femmes (76 %).
Classification (rang B)
Les adénomes peuvent être invasifs en infiltrant les sinus caverneux sur les côtés, ou le plancher sellaire en bas, ou la citerne optochiasmatique, voire le plancher du troisième ventricule en haut.
Mode de découverte
Syndrome tumoral
Les troubles visuels sont liés à une compression du chiasma optique, qui est situé juste au-dessus de l’hypophyse. Les patients peuvent se plaindre d’une sensation de flou visuel, de voile devant les yeux, plus rarement ils remarquent une altération de leur champ de vision (accident de voiture par non-perception d’un autre véhicule arrivant sur le côté).
Trois examens ophtalmologiques sont à réaliser pour diagnostiquer ces troubles visuels :
- l’acuité visuelle, qui est le plus souvent conservée ;
- le classique champ visuel de Goldmann, ou aujourd’hui le champ visuel automatisé, qui peut montrer une quadranopsie bitemporale supérieure, ou une hémianopsie bitemporale si les troubles sont plus sévères. Parfois, les patients ont une hémianopsie latérale homonyme si la tumeur comprime une bandelette optique en arrière du chiasma ;
- le fond d’œil, à la recherche d’une pâleur papillaire, signant une souffrance ancienne du nerf optique et donc un moindre espoir de récupération visuelle après chirurgie.
- des céphalées intenses et brutales ;
- un syndrome méningé ;
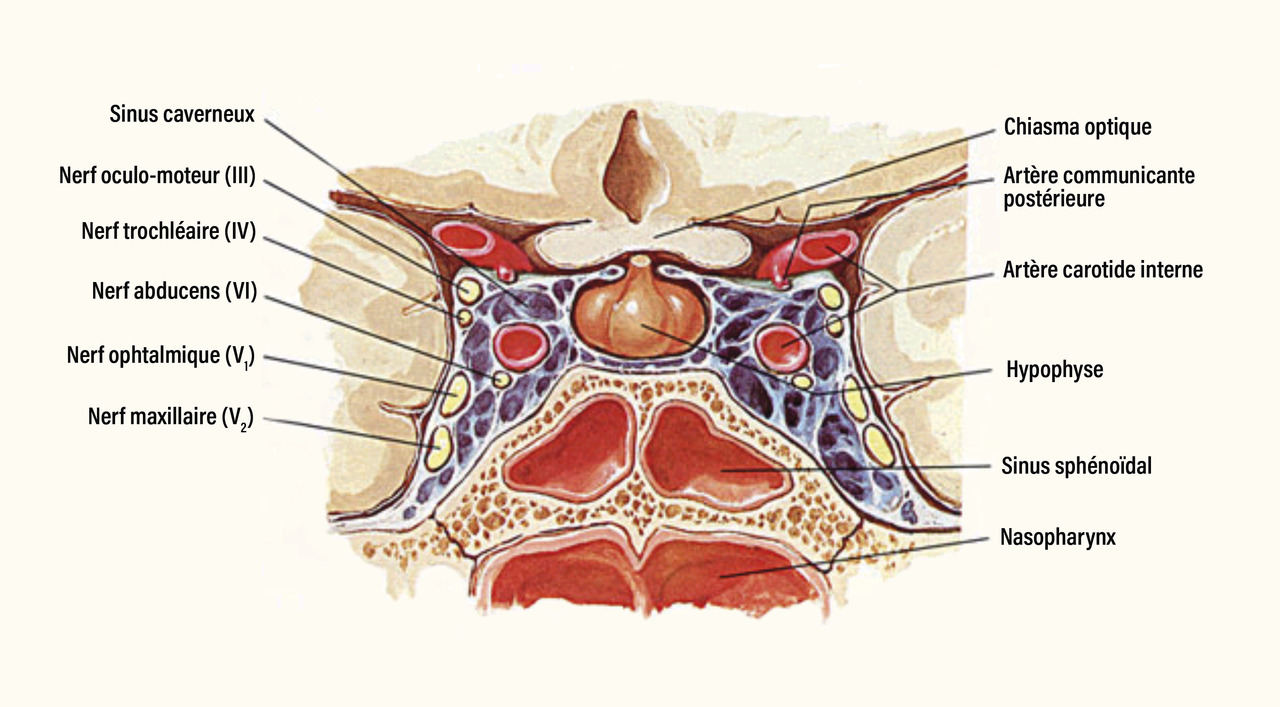
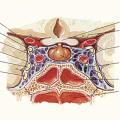
- une paralysie oculomotrice liée à une compression des nerfs crâniens (III, IV et VI) dans le sinus caverneux (fig. 1) ;
- des troubles visuels allant de la baisse de l’acuité visuelle à la cécité ;
- un ptosis (atteinte du III extrinsèque), avec mydriase (atteinte du III intrinsèque) ;
- un syndrome confusionnel, voire un coma ;
- des signes d’insuffisance hypophysaire, notamment corticotrope.
Hypersécrétion
Hyperprolactinémie
Chez la femme
Dans de rares cas, les femmes conservent des cycles mais qui sont le plus souvent anovulatoires : une courbe de température et un dosage de progestérone en deuxième partie de cycle permettent de faire le diagnostic d’anovulation.
Le deuxième signe clinique d’hyperprolactinémie chez la femme est la galactorrhée (
Chez la jeune fille prépubaire, on observe une aménorrhée primaire et un retard pubertaire.
Chez l’homme
Une gynécomastie peut être observée mais n’est pas systématique.
L’enfant ou l’adolescent présente un retard pubertaire ou un arrêt de la puberté.
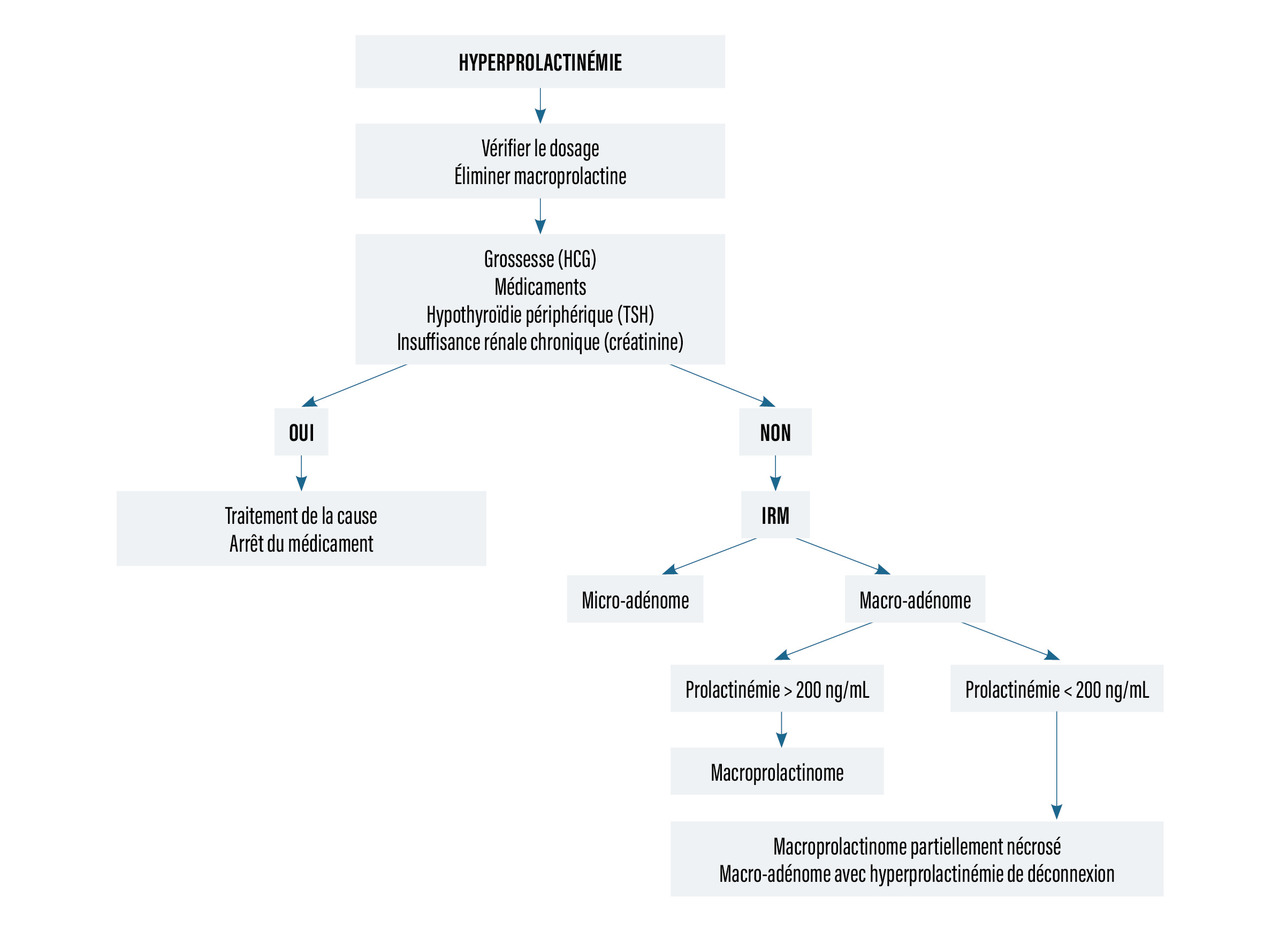
Diagnostic biologique
Diagnostic étiologique (rang B)
- l’hyperœstrogénie de la grossesse ;
- les hyperprolactinémies médicamenteuses : les médicaments inhibant la dopamine élèvent les chiffres de prolactine (neuroleptiques, antidépresseurs, antivomitifs, opiacés). Il est conseillé de vérifier pour chaque médicament pris par le patient la possibilité d’une hyperprolactinémie ;
- l’hypothyroïdie sévère, où l’élévation de la TRH peut stimuler les cellules lactotropes (cause rare d’hyperprolactinémie) ;
- l’insuffisance rénale chronique (diminution de la clairance de la prolactine).
- sécrétion de prolactine par un micro-adénome (prolactinémie < 200 ng/mL) ou par un macro-adénome hypophysaire (prolactinémie > 200 ng/mL) ;
- hyperprolactinémie de déconnexion, liée à une compression de la tige pituitaire, qui entraîne une levée du signal inhibiteur de la dopamine (macro-adénome à l’imagerie par résonance magnétique [IRM] et prolactinémie < 200 ng/mL).
Excès d’hormone de croissance : acromégalie
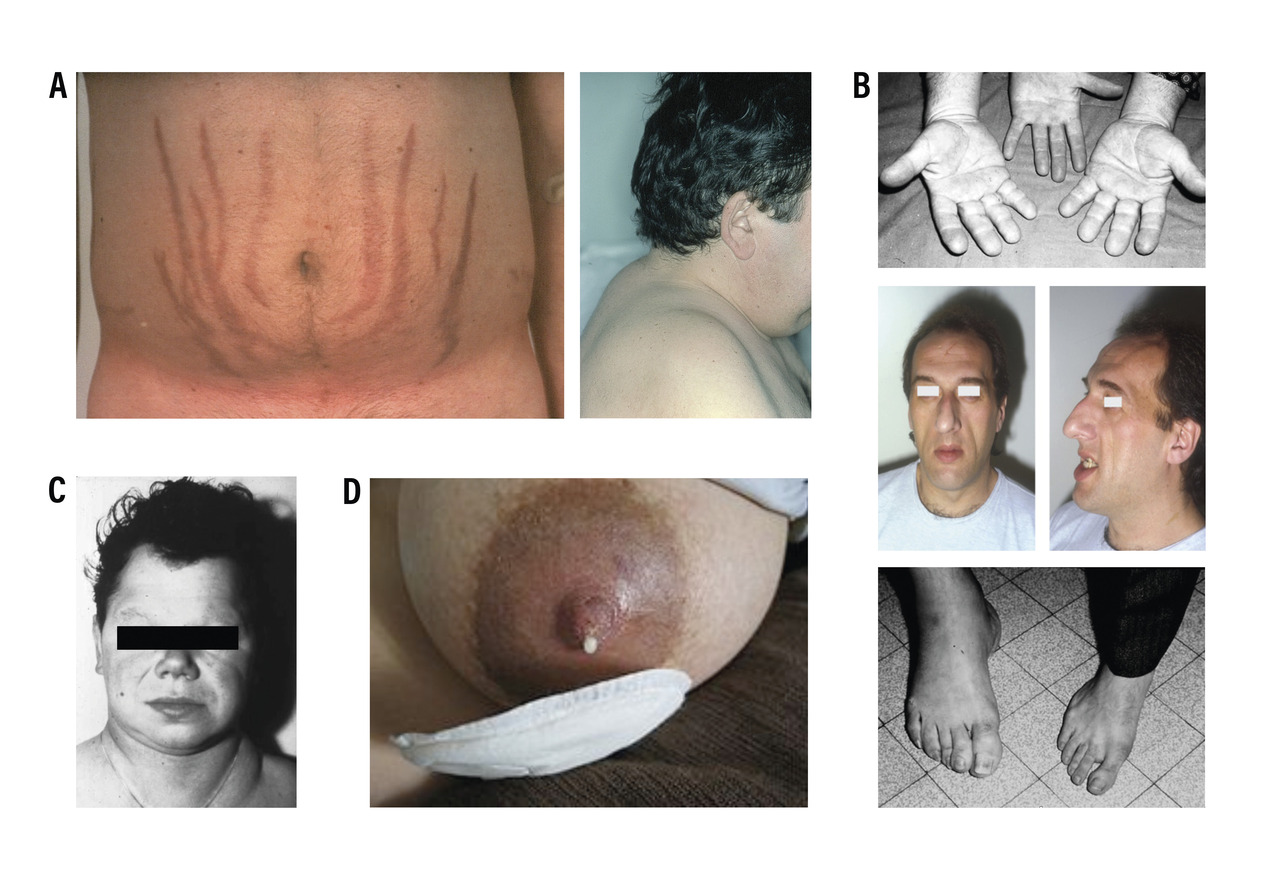
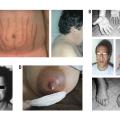
Signes cliniques
En sus de ces déformations, les patients se plaignent de signes généraux tels que des sueurs, surtout nocturnes, des céphalées (quelle que soit la taille de l’adénome), des douleurs articulaires, des paresthésies des mains et une asthénie (
À l’interrogatoire, un ronflement voire des symptômes d’apnées du sommeil doivent être recherchés.
L’hypertension artérielle est présente chez 20 à 50 % des patients.
Diagnostic de l’acromégalie
Chez l’acromégale, la GH sous HGPO reste supérieure à 0,4 ng/mL (1 mUI/L), voire présente une stimulation paradoxale (rang C).
Complications de l’acromégalie
Sur le plan articulaire, les patients présentent des arthralgies mécaniques périphériques et des lombalgies qui évoluent vers l’arthrose à partir de 45 ans.
Le syndrome du canal carpien est très fréquent, lié à un œdème du nerf médian.
Sur le plan cardiovasculaire, l’acromégalie conduit à une augmentation du débit cardiaque, une cardiomégalie, une valvulopathie. L’absence de traitement de l’acromégalie conduit à une insuffisance cardiaque congestive.
Sur le plan métabolique, la GH est hyperglycémiante et induit une insulinorésistance. L’intolérance au glucose et le diabète sont des complications fréquentes de l’acromégalie.
Le syndrome des apnées du sommeil touche 60 à 80 % des acromégales.
L’acromégalie entraîne une organomégalie, avec une augmentation du risque de goitre et le développement de nodules thyroïdiens. La prévalence des polypes coliques est également augmentée.
Bilan d’une acromégalie (rang B)
- les dosages de l’IGF-1 et la sécrétion de GH sous HGPO ;
- l’évaluation du retentissement sur les autres axes hypophysaires et le retentissement visuel en cas de macro-adénome ;
- le bilan tumoral (IRM hypophysaire) ;
- la recherche de complications : échographie cardiaque, échographie thyroïdienne, coloscopie, pression artérielle, signes de syndrome des apnées du sommeil, glycémie.
Adénome corticotrope : hypercorticisme ACTH-dépendant d’origine hypophysaire (maladie de Cushing)
Signes cliniques
Chez la femme, des signes d’hyperandrogénie se développent : hirsutisme, hyperséborrhée, voire acné. Par ailleurs, l’hypercorticisme induit un hypogonadisme hypogonadotrope fonctionnel avec spanioménorrhée, voire aménorrhée, chez la femme et impuissance chez l’homme, ainsi qu’une baisse de la libido.
Sur le plan cardiovasculaire, l’hypertension artérielle est très fréquente,
Sur le plan neuropsychique, l’excès de cortisol peut aggraver ou révéler un trouble psychiatrique latent, des troubles du sommeil, un syndrome dépressif.
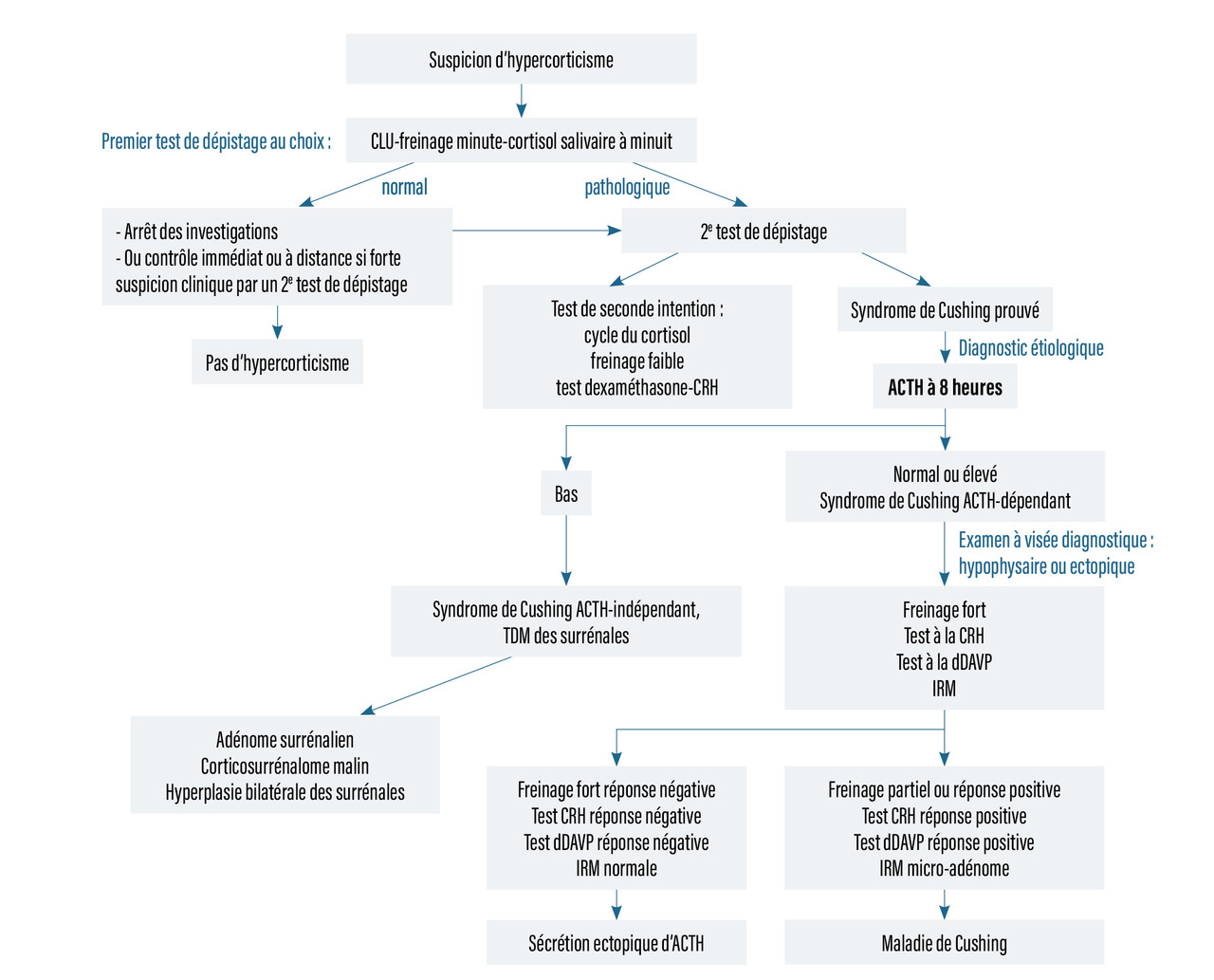
Diagnostic biologique
Dans un premier temps, il est nécessaire d’affirmer une sécrétion excessive de cortisol.
Pour dépister un syndrome de Cushing, un des trois tests possibles en externe est à réaliser en première intention : le dosage du cortisol sur les urines de 24 heures (CLU), le freinage minute ou le cortisol salivaire nocturne (23 h-minuit) ;
- pour le dosage du CLU, un recueil complet des urines de 24 heures est nécessaire, et la créatinine urinaire est dosée pour s’assurer du bon recueil (urines du matin jetées, puis recueil de toutes les urines de la journée et de la nuit jusqu’aux premières urines du lendemain matin) ;
- le freinage minute consiste en la prise de 1 mg de dexaméthasone à 23 h-minuit, et en un dosage le lendemain matin à 8 h du cortisol plasmatique. La dexaméthasone est un glucocorticoïde de synthèse puissant qui n’est pas reconnu quand on dose le cortisol sanguin ou urinaire (on considère que le test est normal [freinage positif] si la cortisolémie est < 1,8 µg/dL [rang C]) ;
- pour le cortisol salivaire, une salivette est à mâcher pendant une minute entre 23 h et minuit, puis à déposer au laboratoire le lendemain matin. Ce dosage n’est pas remboursé. Une valeur basse à minuit – moment où la sécrétion de cortisol est la plus basse – élimine un syndrome de Cushing (cycle nycthéméral conservé) [rang C] ; à l’inverse, une valeur élevée est en faveur du diagnostic d’hypersécrétion (rang C).
Si un test est normal et la suspicion clinique faible, on ne poursuit pas les explorations ; si la suspicion clinique est forte, un deuxième test peut être utilisé dans l’immédiat ou à distance.
Si un test de dépistage est en faveur d’un syndrome de Cushing, un deuxième test de dépistage est réalisé. Si les deux tests de dépistage sont concordants, le syndrome de Cushing est affirmé.
Des examens de deuxième intention sont nécessaires si les deux tests sont discordants :
- cycle nycthéméral du cortisol, la sécrétion étant physiologiquement plus importante le matin à 8 h, avec un nadir vers 23 h-minuit. En cas de syndrome de Cushing, on observe une perte du rythme circadien, avec un cycle dit « plat » ;
- un test de freinage faible ou freinage standard est plus rarement réalisé car les sensibilité et spécificité ne sont pas meilleures que le freinage minute (rang C). Il consiste en un recueil d’urine pendant 48 heures avec prise concomitante de dexaméthasone 0,5 mg toutes les six heures. Un CLU < 10 µg/24 heures élimine le diagnostic de syndrome de Cushing.
Un test dexaméthasone-corticotropin releasing hormone (CRH) permet de distinguer le pseudo-Cushing (rang C).
Anomalies biologiques non spécifiques
L’hémogramme peut objectiver une hyperleucocytose à polynucléaires neutrophiles, du fait d’une démargination des leucocytes (rang C).
Sur le plan endocrinien, l’axe gonadotrope est freiné par l’hypercorticisme (hypogonadisme hypogonadotrope) [rang C].
Diagnostic étiologique (rang B)
- une ACTH effondrée affirme le caractère ACTH-indépendant, autrement dit une origine surrénalienne (adénome ou corticosurrénalome malin) ;
- inversement, une valeur d’ACTH dans la norme ou plus élevée signe le caractère ACTH-dépendant.
- présence d’un micro-adénome à l’IRM ;
- test de freinage fort à la dexaméthasone positif (2 mg de dexaméthasone toutes les 6 heures, pendant 48 heures, recueil des urines le 2e jour et cortisol ACTH de 8 h à la 48e heure ;
- test à la CRH positif ;
- test à la desmopressine (dDAVP) positif (dû à l’expression ectopique de récepteur à la dDAVP sur les cellules adénomateuses corticotropes).
Le principal diagnostic différentiel des syndromes de Cushing ACTH-dépendant est le pseudo-Cushing, par hypercorticisme fonctionnel. Le contexte est souvent évocateur : syndrome dépressif, alcoolisme, psychose, stress intense. Ces situations entraînent une élévation modérée de la sécrétion de cortisol et une résistance relative aux glucocorticoïdes (rang B).
Le test au CRH réalisé après un freinage à la dexaméthasone et le contexte permettent de faire le diagnostic (rang C).
L’arbre diagnostique devant un hypercorticisme clinique est détaillé dans la
Autres hypersécrétions
Adénome gonadotrope : excès de FSH et/ou de LH
Sur le plan biologique, la testostérone ou l’estradiol sont bas alors que les gonadotrophines ne sont pas élevées (dans les valeurs normales ou basses) [rang B].
Le diagnostic d’adénome gonadotrope est le plus souvent histologique, par la démonstration de la présence de gonadotrophines en immunohistochimie (rang C).
Adénome thyréotrope (rang C)
Insuffisance hypophysaire
Signes propres aux déficits de chaque fonction hypophysaire
Signes liés à l’insuffisance corticotrope
Les patients présentent une dépilation axillaire et pubienne liée aux déficits en androgènes surrénaliens.
La zone glomérulée n’est pas touchée par l’insuffisance corticotrope, ainsi le système rénine-angiotensine n’est pas atteint : il n’y a pas d’hyperkaliémie ni de perte de sel comme dans l’insuffisance surrénale, et l’hypotension est rare (rang C). En cas de décompensation corticotrope, on observe une hyponatrémie de dilution liée à la sécrétion inappropriée d’ADH (hormone antidiurétique ou vasopressine) secondaire à la carence en cortisol (rang C).
Signes liés à l’insuffisance gonadotrope
Chez la femme avant l’âge de la ménopause, l’aménorrhée sans bouffées de chaleur est un signe fréquent, associée à une sécheresse et une atrophie des muqueuses vaginales et vulvaire entraînant une dyspareunie.
Signes liés à l’insuffisance thyréotrope
Signes liés à l’insuffisance somatotrope
Chez l’enfant, le déficit en GH entraîne un retard de croissance, une cassure de la courbe de croissance.
Bilan hormonal d’un déficit antéhypophysaire
De façon globale, dans les déficits hypophysaires, les hormones hypophysaires sont normales, et les hormones périphériques basses. Ainsi, un dosage isolé des hormones hypophysaires ne permet pas de faire le diagnostic.Déficit corticotrope
Étant donné les inconvénients de l’hypoglycémie insulinique, d’autres tests peuvent être utilisés comme le test au Synacthène ou le test à la CRH (rang B). Le Synacthène immédiat teste la capacité des surrénales à produire du cortisol (rang B). Après injection de 250 mg de Synacthène (ACTH de synthèse) en intramusculaire, le cortisol est dosé à 30 et 60 minutes. Si le cortisol est supérieur à 180 µg/L (500 nmol/L), il n’y a pas de déficit (rang B).
C’est un mauvais test pour évaluer l’étage hypothalamo-hypophysaire puisque ce test peut être normal (pic > 180 µg/L) en cas d’insuffisance corticotrope partielle et/ou récente (datant de moins de 3 mois) [rang C].
Un simple dosage du cortisol plasmatique à 8 h a une mauvaise performance (rang B). Cependant, une valeur de cortisolémie à 8 h inférieure à 50 µg/L (140 nmol/L) affirme un déficit en cortisol (d’origine centrale ou périphérique) et une valeur supérieure à 135 µg/L (365 nmol/L) l’élimine (rang B).
Lorsque la sécrétion de cortisol est insuffisante, c’est l’ACTH à 8 h qui affirme la cause du déficit :
- hypophysaire (ou central ou insuffisance corticotrope) si l’ACTH à 8 h est normale ou basse ;
- surrénalien (uropériphérique) si l’ACTH à 8 h est élevée.
Déficit somatotrope
L’hypoglycémie insulinique est un test de référence. Elle permet de tester l’axe corticotrope et somatotrope dans le même temps (rang C). Un autre test utilisé est la GHRH couplée à l’arginine (growth hormone-releasing hormone) [rang C].
Déficit thyréotrope (rang B)
Déficit gonadotrope (rang B)
Déficit lactotrope (rang B)
Diabète insipide
Imagerie
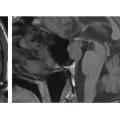
IRM hypophysaire
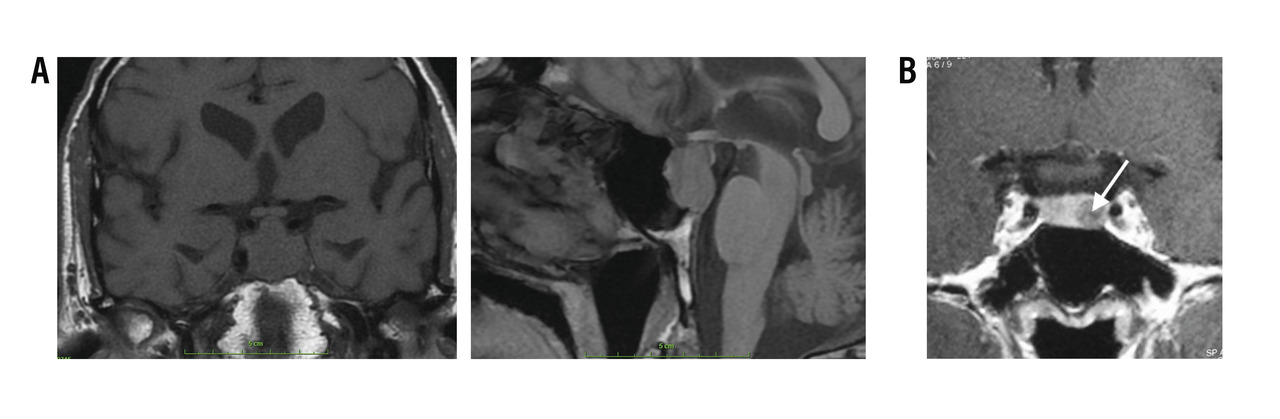
Les micro-adénomes (< 10 mm) sont intrasellaires, ils apparaissent arrondis, homogènes (rang B), en hyposignal en T1 et hypo-, iso- ou hypersignal en T2 (rang C). La prise de contraste est homogène, volontiers retardée par rapport au parenchyme sain (
Les macro-adénomes (> 10 mm) s’étendent en extrasellaire (rang B), leur signal est iso-T1, et souvent hétérogène en T2, les plages nécrotiques apparaissent en hypersignal T2 (rang C). La prise de contraste est le plus souvent hétérogène hypo-intense par rapport à l’hypophyse saine (
Diagnostics différentiels en imagerie
Le craniopharyngiome apparaît en IRM comme une masse hétérogène, souvent kystique, avec une capsule prenant le contraste (rang C). Les calcifications sont fréquentes et nécessitent de réaliser un scanner en fenêtre osseuse car elles sont très mal visualisées en IRM (rang C).
Les méningiomes sont des lésions qui prennent très fortement le produit de contraste et qui « s’accrochent » à la dure-mère, donnant une image en « queue de comète » (rang C).
Les autres diagnostics différentiels sont la grosse hypophyse de la femme jeune, l’hypophysite auto-immune, les métastases, la sarcoïdose, l’histiocytose, la tuberculose (rang C).
Conclusion
POINTS FORTS À RETENIR
Le syndrome tumoral (céphalées et troubles visuels) est lié à une extension suprasellaire d’un macro-adénome hypophysaire (> 10 mm), avec compression du chiasma par l’adénome ; il impose une étude du champ visuel (hémianopsie bitemporale).
L’IRM hypophysaire est l’examen de référence.
Les micro-adénomes (< 10 mm) posent le problème d’une hypersécrétion, qui est à rechercher par l’examen clinique et le bilan hormonal. L’hypersécrétion corticotrope ou somatotrope nécessite des tests dynamiques de freinage pour affirmer l’hypersécrétion.
Devant un macro-adénome hypophysaire, une insuffisance hypophysaire doit être recherchée. Le diagnostic biologique de l’insuffisance hypophysaire impose un dosage des hormones périphériques qui sont abaissées alors que les hormones hypophysaires sont normales. Un bilan dynamique est nécessaire pour les axes corticotrope et somatotrope.
La découverte d’un syndrome de Cushing (dépistage de première intention par cortisol libre urinaire ou cortisol salivaire à minuit ou freinage minute, test de deuxième intention par le cycle du cortisol ou le freinage faible à la dexaméthasone) impose un diagnostic étiologique reposant sur l’ACTH à 8 h dans un premier temps. Si celui-ci est normal ou élevé, d’autres tests distinguent la maladie de Cushing (adénome corticotrope) de la sécrétion ectopique d’ACTH (d’origine tumorale).
L’hyperprolactinémie doit être confirmée par un dosage en laboratoire spécialisé, elle peut être secondaire à une prise médicamenteuse (psychotropes) même si elle est le plus souvent liée à un adénome à prolactine. La sécrétion est proportionnelle à la taille. Ainsi une valeur de prolactinémie peu élevée (< 200 ng/mL) en regard d’un macro-adénome fait évoquer une hyperprolactinémie de déconnexion.
Un adénome hypophysaire ne s’accompagne pas de diabète insipide.
 Encadrés
Encadrés