L’enjeu principal est le diagnostic précoce qui doit être évoqué devant une symptomatologie souvent aspécifique, surtout cardiaque ou rénale, non expliquée chez des patients souvent âgés et atteints d’une gammapathie monoclonale.
L’amylose est une maladie causée par le dépôt de protéines ayant acquis la capacité de polymériser entre elles sous forme de fibrilles plissées, résistantes aux mécanismes de dégradation et de remaniements normalement présents dans les tissus. Ces dépôts extra-cellulaires sont principalement localisés dans la paroi des vaisseaux sanguins et sur les membranes basales, entraînant une désorganisation des tissus aboutissant à une dysfonction d’organe. L’amylose AL, la plus fréquente dans les pays industrialisés en dehors de l’amylose cardiaque par dépôts de transthyrétine non mutée (amylose sénile), est liée à des dépôts de chaînes légères libres d’immunoglobulines monoclonales et s’inscrit dans le cadre plus large d’un groupe de maladies appelées « gammapathies monoclonales de signification clinique ».1 Ces immunoglobulines monoclonales sont sécrétées par des clones plasmocytaires ou lymphocytaires B, et peuvent être responsables de lésions organiques diverses et potentiellement graves, dont l’amylose AL. Dans ce cas, la prolifération est plasmocytaire dans plus de 90 % des cas (avec une immunoglobuline [Ig] monoclonale d’isotype IgG, IgA ou dans 50 % des cas uniquement des chaînes légères libres). Dans les 10 % des cas restant, il s’agit d’un clone lympho-plasmocytaire (sécrétant plutôt une IgM).
Épidémiologie
Cette pathologie est rare mais non exceptionnelle. On estime en France l’incidence à 600 à 700 nouveaux cas par an. Sa prévalence a nettement augmenté depuis 20 ans du fait d’une amélioration des procédures diagnostiques et d’une meilleure survie des patients. L’âge moyen au diagnostic est de 65 ans.
Diagnostic
L’amylose AL peut être primitive, le plus souvent, ou secondaire, associée à un myélome symptomatique ou un lymphome.
Symptomatologie clinique
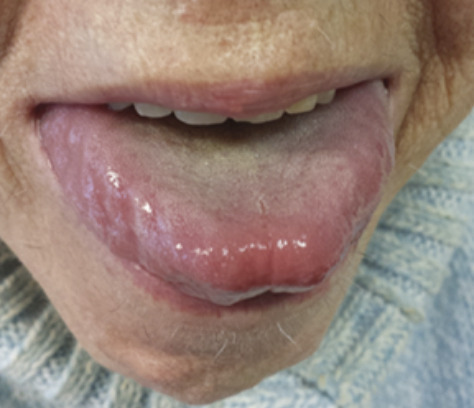
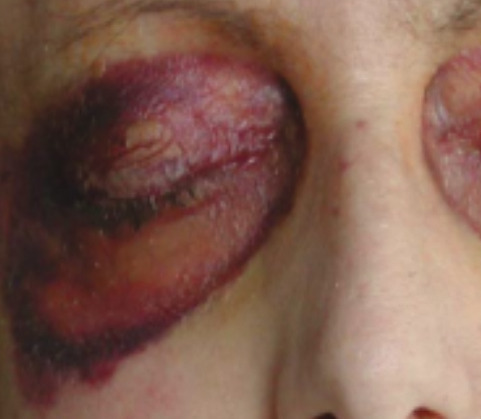
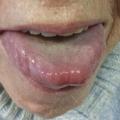
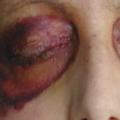
Les signes cliniques les plus évocateurs sont les hématomes cutanés, en particulier des paupières, et la macroglossie, mais le diagnostic est souvent retardé devant une symptomatologie fréquemment non spécifique (tableau 1 ).
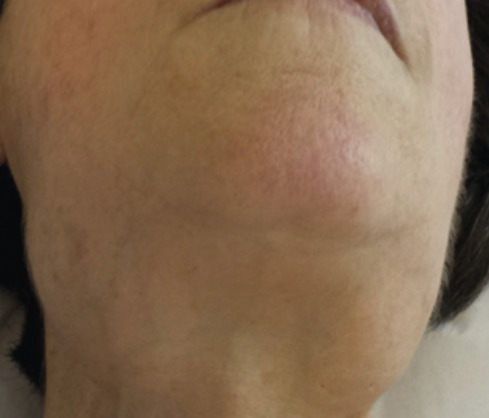
L’amylose AL peut affecter tous les organes excepté le système nerveux central (tableau 1 et fig. 1 à 3 ). L’atteinte peut être localisée à un seul organe ou systémique.
Il faut y penser devant l’altération de la fonction d’un organe qui n’est pas expliquée, en particulier chez des patients porteurs d’une gammapathie monoclonale de signification indéterminée (MGUS, pour monoclonal gammopathy of undetermined significance).
Les deux atteintes les plus fréquentes sont l’atteinte rénale se traduisant le plus souvent par un syndrome néphrotique responsable d’œdèmes des membres inférieurs, et l’atteinte cardiaque restrictive responsable d’une asthénie et d’une dyspnée d’effort et souvent de troubles de la conduction ou du rythme cardiaque et de syncopes. Le diagnostic doit également être évoqué, en particulier chez un patient ayant une MGUS, devant un syndrome du canal carpien, surtout bilatéral, une neuropathie périphérique surtout associée à une dysautonomie ou une hépatomégalie inexpliquée. Les signes cliniques et paracliniques des différentes atteintes sont résumés dans le tableau 1 .
L’amylose AL peut affecter tous les organes excepté le système nerveux central (
Il faut y penser devant l’altération de la fonction d’un organe qui n’est pas expliquée, en particulier chez des patients porteurs d’une gammapathie monoclonale de signification indéterminée (MGUS, pour monoclonal gammopathy of undetermined significance).
Les deux atteintes les plus fréquentes sont l’atteinte rénale se traduisant le plus souvent par un syndrome néphrotique responsable d’œdèmes des membres inférieurs, et l’atteinte cardiaque restrictive responsable d’une asthénie et d’une dyspnée d’effort et souvent de troubles de la conduction ou du rythme cardiaque et de syncopes. Le diagnostic doit également être évoqué, en particulier chez un patient ayant une MGUS, devant un syndrome du canal carpien, surtout bilatéral, une neuropathie périphérique surtout associée à une dysautonomie ou une hépatomégalie inexpliquée. Les signes cliniques et paracliniques des différentes atteintes sont résumés dans le
Diagnostic biologique et anatomopathologique
Les examens d’orientation initiaux comprennent la recherche d’une MGUS et d’un excès de chaînes légères libres monoclonales circulantes kappa ou lambda dans le sérum par le dosage des chaînes légères libres sériques. Un algorithme diagnostique a été proposé en 2018.2
Le diagnostic de certitude est l’anatomopathologie. En première intention les biopsies non invasives sont indiquées (biopsie de glandes salivaires accessoires ou biopsie de la graisse sous-cutanée). Si les résultats sont négatifs pour l’amylose et que la suspicion est forte, une biopsie de l’organe atteint est nécessaire. Le diagnostic est confirmé par la présence de dépôts fibrillaires amorphes, acellulaires au contact des matrices extra-cellulaires positifs en coloration rouge congo avec une biréfringence dichromique jaune-verte en lumière polarisée. Enfin, il est absolument indispensable de caractériser la protéine amyloïde par une technique d’immunomarquage sur du tissu congelé, afin de mettre en évidence la chaîne légère monoclonale.
Après confirmation anatomopathologique de l’amylose AL, la réalisation d’un myélogramme est nécessaire. Il trouve le plus souvent une population plasmocytaire monoclonale, l’infiltration médullaire est habituellement faible ; 40 % des patients ont tout de même plus de 10 % de plasmocytes sur l’évaluation médullaire et donc un diagnostic de myélome indolent associé. Dans ce cas, l’évolution vers un myélome symptomatique est rare. Une étude cytogénétique est utile, en particulier à la recherche d’une translocation (11;14) présente dans 50 % des cas et dont la présence oriente les décisions thérapeutiques.
Le diagnostic de certitude est l’anatomopathologie. En première intention les biopsies non invasives sont indiquées (biopsie de glandes salivaires accessoires ou biopsie de la graisse sous-cutanée). Si les résultats sont négatifs pour l’amylose et que la suspicion est forte, une biopsie de l’organe atteint est nécessaire. Le diagnostic est confirmé par la présence de dépôts fibrillaires amorphes, acellulaires au contact des matrices extra-cellulaires positifs en coloration rouge congo avec une biréfringence dichromique jaune-verte en lumière polarisée. Enfin, il est absolument indispensable de caractériser la protéine amyloïde par une technique d’immunomarquage sur du tissu congelé, afin de mettre en évidence la chaîne légère monoclonale.
Après confirmation anatomopathologique de l’amylose AL, la réalisation d’un myélogramme est nécessaire. Il trouve le plus souvent une population plasmocytaire monoclonale, l’infiltration médullaire est habituellement faible ; 40 % des patients ont tout de même plus de 10 % de plasmocytes sur l’évaluation médullaire et donc un diagnostic de myélome indolent associé. Dans ce cas, l’évolution vers un myélome symptomatique est rare. Une étude cytogénétique est utile, en particulier à la recherche d’une translocation (11;14) présente dans 50 % des cas et dont la présence oriente les décisions thérapeutiques.
Cardiopathie amyloïde
Le diagnostic d’atteinte cardiaque dans l’amylose AL repose sur l’existence de deux critères : une augmentation de l’épaisseur myocardique supérieure à 12 mm en l’absence d’autre cause et une élévation des marqueurs cardiaques (N-Terminal pro-Brain Natriuretic [NT-proBNP] ou BNP).3, 4
Les signes cliniques devant alerter sont : des signes d’insuffisance cardiaque gauche et/ou droite, l’existence d’une syncope ou lipothymie (en rapport avec une hypotension orthostatique, un trouble de conduction), une pression artérielle le plus souvent abaissée. L’électrocardiogramme est très souvent anormal (environ 90 % des cas) avec un microvoltage, l’existence de pseudo-ondes q de nécrose dans les dérivations antérieures, un bloc auriculo-ventriculaire de type 1, une fibrillation atriale. L’élévation des biomarqueurs cardiaques est un élément important en faveur du diagnostic et leurs valeurs sont pronostiques.5
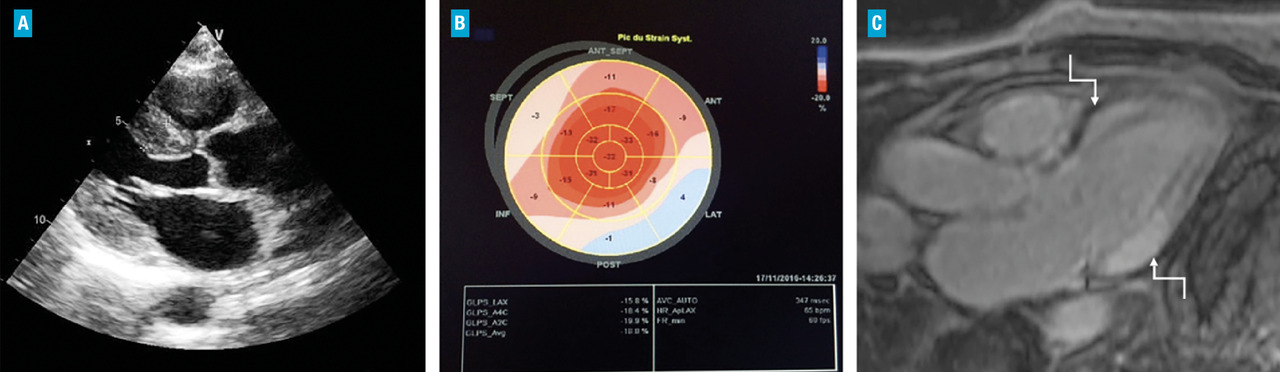
L’échocardiographie est l’examen de première intention en cas de suspicion d’atteinte cardiaque dans l’amylose. Les signes évocateurs sont une hypertrophie ventriculaire gauche concentrique, un ventricule gauche non dilaté voire réduit de taille, une hypertrophie ventriculaire droite, une fraction d’éjection ventriculaire gauche souvent normale contrastant avec une altération de la fonction longitudinale ventriculaire gauche (strain) avec une altération plus importante des segments basaux et un aspect typique en cocarde avec respect de la pointe (fig. 4A et 4B ). On note également un effondrement des vitesses tissulaires avec un flux mitral restrictif, une dilatation des oreillettes et un volume d’éjection systolique abaissé. Un épanchement péricardique de faible abondance est trouvé dans environ 50 % des cas.6
L’imagerie par résonance magnétique (IRM) cardiaque est un examen de deuxième intention, permettant d’apporter en plus des anomalies morphologiques, des éléments de caractérisation tissulaire très informatifs. On peut en effet mesurer le T1 et le volume extracellulaire qui sont élevés en cas de cardiomyopathie amyloïde. De plus, les séquences post-injection de gadolinium peuvent mettre en évidence un rehaussement tardif diffus de topographie non coronarienne en rapport avec des dépôts amyloïdes intramyocardiques (fig. 4C ).7
Pour faire la distinction entre amylose cardiaque AL ou amylose cardiaque par dépôt de transthyrétine, pathologie beaucoup plus fréquente chez la personne âgée, surtout chez les hommes, il est utile de réaliser une scintigraphie osseuse au 99m Tc-PYP qui montre une fixation cardiaque constante en cas d’amylose à transthyrétine et beaucoup plus rare dans les amyloses AL.
Les signes cliniques devant alerter sont : des signes d’insuffisance cardiaque gauche et/ou droite, l’existence d’une syncope ou lipothymie (en rapport avec une hypotension orthostatique, un trouble de conduction), une pression artérielle le plus souvent abaissée. L’électrocardiogramme est très souvent anormal (environ 90 % des cas) avec un microvoltage, l’existence de pseudo-ondes q de nécrose dans les dérivations antérieures, un bloc auriculo-ventriculaire de type 1, une fibrillation atriale. L’élévation des biomarqueurs cardiaques est un élément important en faveur du diagnostic et leurs valeurs sont pronostiques.5
L’échocardiographie est l’examen de première intention en cas de suspicion d’atteinte cardiaque dans l’amylose. Les signes évocateurs sont une hypertrophie ventriculaire gauche concentrique, un ventricule gauche non dilaté voire réduit de taille, une hypertrophie ventriculaire droite, une fraction d’éjection ventriculaire gauche souvent normale contrastant avec une altération de la fonction longitudinale ventriculaire gauche (strain) avec une altération plus importante des segments basaux et un aspect typique en cocarde avec respect de la pointe (
L’imagerie par résonance magnétique (IRM) cardiaque est un examen de deuxième intention, permettant d’apporter en plus des anomalies morphologiques, des éléments de caractérisation tissulaire très informatifs. On peut en effet mesurer le T1 et le volume extracellulaire qui sont élevés en cas de cardiomyopathie amyloïde. De plus, les séquences post-injection de gadolinium peuvent mettre en évidence un rehaussement tardif diffus de topographie non coronarienne en rapport avec des dépôts amyloïdes intramyocardiques (
Pour faire la distinction entre amylose cardiaque AL ou amylose cardiaque par dépôt de transthyrétine, pathologie beaucoup plus fréquente chez la personne âgée, surtout chez les hommes, il est utile de réaliser une scintigraphie osseuse au 99m Tc-PYP qui montre une fixation cardiaque constante en cas d’amylose à transthyrétine et beaucoup plus rare dans les amyloses AL.
Pronostic
L’urgence est d’estimer la gravité de l’atteinte cardiaque qui conditionne le pronostic à court terme. Depuis 2004, il existe une classification pronostique, dépendante de la sévérité de cette atteinte qui s’appuie sur les deux marqueurs sériques, la troponine cardiaque et le NT-proBNP. Des seuils sériques de troponine cardiaque à 0,035 μg/L et NT-proBNP à 332 ng/L (ou BNP > 81 pg/L) ont été retenus pour déterminer trois stades de gravité (classification de la Mayo Clinic) : 1. les deux marqueurs en dessous du seuil ; 2. un des deux marqueurs au-dessus du seuil ; et 3. les deux marqueurs au-dessus du seuil avec une différence statistiquement significative de médiane de survie de 26,4, 10,5, et 3,5 mois respectivement (p < 0,001).8 Cette classification a été complétée par une étude en 2013 pour les patients les plus graves (stade 3), qui a démontré qu’un dosage sérique de NT-proBNP supérieur à 8 500 ng/L impactait négativement la survie globale, en définissant ainsi un sous-groupe de patients avec un pronostic très sombre et une médiane de survie globale de 3 mois seulement (stade 3b).9
Traitement causal
L’objectif principal est de diminuer rapidement la production de la chaîne légère monoclonale toxique en ciblant le clone plasmocytaire ou lymphocytaire responsable de la sécrétion. Les thérapeutiques dérivent donc naturellement de celles utilisées dans le myélome multiple ou les lymphomes. Cependant, en cas d’amylose AL primitive, les protocoles proposés sont moins intensifs et de plus courte durée. L’association de différents médicaments est préconisée (inhibiteurs du protéasome, alkylants, immunomodulateurs, corticoïdes, anticorps monoclonaux anti-CD38 ou anti-CD20), l’intensité du traitement dépendant de la sévérité de l’atteinte cardiaque et de la nature de l’hémopathie responsable. Plus la maladie est grave, plus rapidement il faut obtenir une réponse hématologique. Il faut moduler le traitement en monitorant régulièrement les chaînes légères libres circulantes afin d’adapter rapidement le traitement pour obtenir une décroissance rapide. Les recommandations du centre de référence national de l’amylose AL et des maladies de dépôts d’immunoglobulines monoclonales sont disponibles en ligne.10 Les associations thérapeutiques sont privilégiées, sous forme de bithérapie pour les stades 1, et trithérapie pour les stades 2 et 3. Elles comprennent des alkylants type melphalan ou cyclophosphamide en association à la dexaméthasone en systématique, et du bortézomib pour les stades 2 et 3. Des thérapeutiques plus récentes sont actuellement en cours d’étude dans l’amylose AL tels que les inhibiteurs de BCL2, ainsi que des anticorps anti-dépôts qui permettraient d’accélérer la clairance des dépôts en association concomitante avec le traitement causal.
Surveillance de l’efficacité des traitements
L’efficacité des thérapeutiques s’évalue dans un premier temps sur la décroissance de la différence entre chaînes légères impliquées et non impliquées (dFLC). Quatre grades de réponses ont été définis ayant un impact en termes de survie globale : réponse complète (ratio de chaînes légères normal et immunofixations sérique et urinaire normales), très bonne réponse partielle (dFLC < 40 mg/L), réponse partielle (diminution de la dFLC > 50 %) et non répondeur.11 En effet plus la réponse hématologique est profonde, plus la médiane de survie globale augmente et de nouveaux critères sont en cours de validation telle l’obtention d’une dFLC inférieure à 10 mg/L comme objectif du traitement.12
Par ailleurs, il est important d’évaluer la réponse d’organe définie par plusieurs études. Notamment l’évolution des marqueurs sériques NT-proBNP et troponine cardiaque qui ont un impact sur la survie globale ainsi que la créatininémie ou la protéinurie important pour le pronostic rénal (tableau 2 ).13, 14
Par ailleurs, il est important d’évaluer la réponse d’organe définie par plusieurs études. Notamment l’évolution des marqueurs sériques NT-proBNP et troponine cardiaque qui ont un impact sur la survie globale ainsi que la créatininémie ou la protéinurie important pour le pronostic rénal (
Traitement symptomatique
Les soins de support des atteintes d’organes sont indispensables car le délai avant l’amélioration de l’atteinte d’organe peut être long. Les recommandations des autres collèges de spécialités ne s’appliquent pas forcément aux atteintes d’organe induites par l’amylose AL. En effet, l’insuffisance cardiaque secondaire dans ce contexte peut être aggravée par les inhibiteurs de l’enzyme de conversion aggravant l’hypotension et surtout les bêtabloquants potentiellement mal tolérés sur le plan hémodynamique dû à une faible réserve inotrope. Le traitement de la congestion repose sur l’utilisation des diurétiques de l’anse +/- associés aux antagonistes des récepteurs aux minéralocorticoïdes si la fonction rénale le permet. De plus, il faut discuter de façon collégiale assez précocement de l’implantation de dispositifs implantables type stimulateur cardiaque (pacemaker) en cas de troubles de la conduction cardiaque. L’utilisation d’anticoagulants pour les troubles du rythme cardiaque ou les syndromes néphrotiques avec hypo-albuminémie sont également à discuter avec les spécialistes devant une susceptibilité hémorragique accrue chez ces patients. Il existe en effet dans environ 10 % des cas un déficit en facteur X et une fibrinolyse accrue.
Enfin, il ne faut pas oublier l’utilisation des prophylaxies anti-infectieuse accompagnant éventuellement les traitements qui sont immunosuppresseurs (sulfaméthoxazole si dexaméthasone, Zelitrex si bortézomib et Oracilline ou Clamoxyl en prévention des infections bactériennes) ainsi que la mise à jour du calendrier vaccinal.
Enfin, il ne faut pas oublier l’utilisation des prophylaxies anti-infectieuse accompagnant éventuellement les traitements qui sont immunosuppresseurs (sulfaméthoxazole si dexaméthasone, Zelitrex si bortézomib et Oracilline ou Clamoxyl en prévention des infections bactériennes) ainsi que la mise à jour du calendrier vaccinal.
Un pronostic nettement amélioré
c’est une maladie au pronostic péjoratif avec une médiane de survie courte en l’absence de prise en charge spécifique. En 1995, la médiane de survie globale était estimée à 13 mois. Ce pronostic s’est nettement amélioré depuis les années 2 000 même si la mortalité reste élevée pour les patients avec une atteinte cardiaque sévère lors du diagnostic dont la médiane de survie est d’environ 7,1 mois actuellement. Pour les autres patients le pronostic est maintenant relativement bon avec des médianes de survie supérieures à 5 ans.
Références
1. Fermand JP, Bridoux F, Dispenzieri A, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood 2018;132:1478-85.
2. Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm. Blood Cancer J 2018;8:44.
3. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol 2005;79:319-28.
4. Lilleness B, Ruberg FL, Mussinelli R, Doros G, Sanchorawala V. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood 2019;133:215-23.
5. Palladini G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003;107:2440–5.
6. Nicol M, Baudet M, Brun S, et al. Diagnostic score of cardiac involvement in AL amyloidosis. Eur Heart J Cardiovasc Imaging 2020;21:542-8.
7. Fontana M, Chung R, Hawkins PN, et al. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev 2015;20:133-44.
8. Dispenzieri A, Gertz MA, Kyle R, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol 2004;22:3751-7.
9. Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood 2013;121:3420-7.
10. https://www. unilim.fr/cr-amylose-al/medecins/prise-en-charge/traitement_amylose/
11. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012;30:4541-9.
12. Manwani R, Cohen O, Sharpley F, et al. A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. Blood 2019;134:2271-80.
13. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am J Hematol 2005;79:319-28.
14. Palladini G, Hegenbart U, Milani P, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood 2014;124:2325-32.
2. Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm. Blood Cancer J 2018;8:44.
3. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol 2005;79:319-28.
4. Lilleness B, Ruberg FL, Mussinelli R, Doros G, Sanchorawala V. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood 2019;133:215-23.
5. Palladini G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003;107:2440–5.
6. Nicol M, Baudet M, Brun S, et al. Diagnostic score of cardiac involvement in AL amyloidosis. Eur Heart J Cardiovasc Imaging 2020;21:542-8.
7. Fontana M, Chung R, Hawkins PN, et al. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev 2015;20:133-44.
8. Dispenzieri A, Gertz MA, Kyle R, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol 2004;22:3751-7.
9. Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood 2013;121:3420-7.
10. https://www. unilim.fr/cr-amylose-al/medecins/prise-en-charge/traitement_amylose/
11. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012;30:4541-9.
12. Manwani R, Cohen O, Sharpley F, et al. A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. Blood 2019;134:2271-80.
13. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am J Hematol 2005;79:319-28.
14. Palladini G, Hegenbart U, Milani P, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood 2014;124:2325-32.
Dans cet article


Résumé
L’amylose AL est une hémopathie rare, caractérisée par des dépôts de chaînes légères d’immunoglobulines, pouvant survenir dans presque tous les organes et pouvant entraîner leur défaillance. L’enjeu principal est le diagnostic précoce qui doit être évoqué devant une symptomatologie souvent aspécifique, surtout cardiaque ou rénale, non expliquée chez des patients fréquemment âgés et porteurs d’une gammapathie monoclonale. Des biopsies non invasives doivent rapidement permettre la confirmation histologique révélant des dépôts positifs au rouge congo et biréfringent jaune-vert en lumière polarisée spécifique de l’amylose. La gravité est évaluée par les marqueurs biologiques d’atteinte cardiaque. Le traitement consiste à éliminer le clone plasmocytaire ou lymphoplasmocytaire sécrétant la chaîne légère amyloïdogène, et à proposer des soins de support spécifiques à cette pathologie.