objectifs
Argumenter les principales hypothèses diagnostiques et justifier les examens complémentaires pertinents.
Argumenter l’attitude thérapeutique dans les anémies carentielles et planifier leur suivi.
Introduction
Définition
L’anémie est définie par une baisse de l’hémoglobine (Hb) en dessous de seuils prédéfinis qui varient en fonction du sexe et de l’âge :
- homme : Hb < 13 g/dL ;
- femme : Hb < 12 g/dL ;
- femme enceinte < 10,5 g/dL.
En plus du taux d’hémoglobine, plusieurs paramètres caractérisent la lignée érythrocytaire :
- le nombre d’hématies ou érythrocytes (normes chez l’homme : de 4 à 6 x109/mm3 ; chez la femme : de 4 à 5 x 109/mm3) ;
- l’hématocrite (normes chez l’homme : de 40 à 54 % ; chez la femme : de 37 à 45 %) ;
- le volume globulaire moyen (VGM = hématocrite/nombre d’hématies) [normale de 80 à 100 fl] ;
- la teneur corpusculaire moyenne en hémoglobine (TCMH = Hb/nombre d’hématies) [normale de 27 à 33 pg/cellule] ;
- la concentration corpusculaire moyenne en hémoglobine (CCMH = Hb/hématocrite) [normale de 32 à 35 pg/cellule].
Signes cliniques
Au niveau général : asthénie.
Au niveau cutanéomuqueux : pâleur cutanéomuqueuse (examen des conjonctivites, pulpe des doigts).
Aux niveaux cardiaque et pulmonaire : dyspnée, tachycardie, souffle valvulaire fonctionnel, angor pouvant se compliquer d’infarctus du myocarde, insuffisance cardiaque.
Au niveau neurologique : acouphènes, vertiges, céphalées.
Tolérance
La tolérance de l’anémie est variable en fonction de sa rapidité d’installation. Ainsi une anémie aiguë (hémolyse, hémorragie) sera associée à des symptômes plutôt sévères et brutaux. En revanche, une anémie chronique (carentielle), même avec des taux d’hémoglobine extrêmement bas, peut être associée à des symptômes cliniques modérés.
Par ailleurs, la tolérance est aussi variable en fonction du terrain du patient. Les patients présentant une certaine fragilité auront un tableau clinique bruyant pour des taux d’hémoglobine modérés (femme enceinte, personne âgée, insuffisant cardiaque).
Traitements
Le traitement de l’anémie est avant tout celui de la cause. La transfusion de culot érythrocytaire est nécessaire avant tout en fonction de la tolérance, bien qu’il existe des seuils transfusionnels. Dans tout bilan d’anémie il faut réaliser un bilan prétransfusionnel avec groupage ABO, Rhésus et RAI.
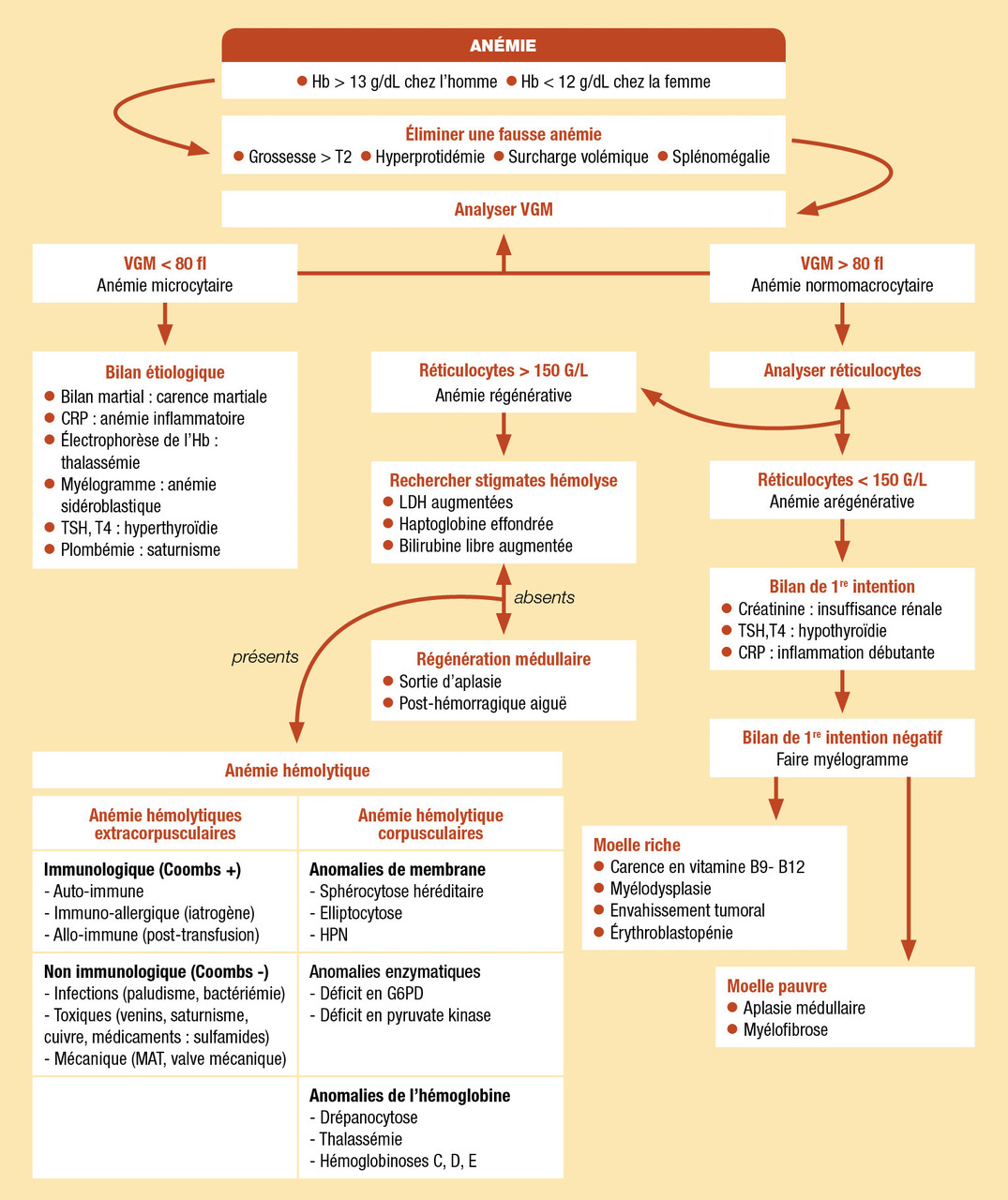
Orientation diagnostique
Éliminer une fausse anémie
Avant de se lancer dans les explorations d’une anémie, il faut avant tout vérifier l’absence d’une fausse anémie par hémodilution : grossesse à partir du 2e trimestre, hypoprotidémie (immunoglobuline monoclonale, notamment IgM), surcharge volémique (insuffisance cardiaque, rénale ou hépatocellulaire) et splénomégalie.
Interpréter le volume globulaire moyen et réticulocytes
Pour le bilan étiologique, on distingue trois types d’anémie distincts :
- microcytaire VGM < 80 fl ;
- normocytaire ou macrocytaire VGM > 80 fl, arégénérative (réticulocytes < 150 G/L) ;
- normocytaire ou macrocytaire VGM > 80 fl, régénérative (réticulocytes > 150 G/L).
Anémie microcytaire
Une anémie microcytaire est systématiquement due à une synthèse insuffisance de l’hémoglobine, soit par défaut de fer dans le plasma, soit par anomalie de la globine. Le dosage des réticulocytes n’est pas forcément nécessaire dans ce contexte, les anémies microcytaires carentielle ou inflammatoire sont arégénératives (cela n’est pas le cas des thalassémies).
Carence martiale
Clinique
C’est la première cause d’anémie dans les pays industrialisés.
Il s’agit d’une anémie chronique, avec un tableau clinique d’installation progressive. En plus du syndrome anémique classique, on retrouve souvent des caractéristiques cliniques évocatrices de syndrome carentiel : fragilité des phanères (chute des cheveux, ongles cassants), fragilité des muqueuses (glossite, œsophagite) et possibles troubles du comportement alimentaire (la maladie de Pica : ingestion de substances non alimentaires comme la terre).
Démarche diagnostique
L’hémogramme retrouve une anémie microcytaire hypochrome pouvant être associée à une thrombocytose réactionnelle. Le frottis sanguin retrouve une anisocytose.
Le bilan martial, à la recherche d’une carence martiale, consiste (selon la Haute Autorité de santé [HAS] 2011) :
- au dosage de la ferritine (abaissée en cas de carence martiale ; cependant, une ferritine normale n’exclut pas une carence martiale en cas de syndrome inflammatoire associé) ;
- ou à l’association de fer sérique abaissé avec l’un des critères suivants :
o transferrine augmentée,
o coefficient de saturation de la transferrine diminué,
o capacité totale de fixation de la transferrine augmentée.
En cas de carence martiale, l’évolution des différents paramètres biologiques n’est pas simultanée : initialement diminution de la ferritine, puis augmentation de la capacité totale de fixation de la transferrine, puis diminution du fer sérique et du coefficient de saturation de la transferrine, puis apparition de la microcytose, puis de l’anémie et, enfin, de l’hypochromie.
Bilan étiologique
Après affirmation de la carence martiale, l’enjeu est d’en définir l’étiologie.
Il existe 3 grands mécanismes de carence martiale :
- pertes de fer par saignement chronique : saignements gynécologiques (examen gynécologique obligatoire chez toutes les femmes : règles abondantes, stérilet, endométriose chez la femme jeune, fibrome et cancer de l’endomètre chez les femmes ménopausées), saignements digestifs (œsophage[varices] ; estomac [ulcère, cancer, gastrite à Helicobacter pylori] ; grêle[maladie de Crohn, maladie cœliaque, polypes] ; côlon [ulcérations, rectocolite hémorragique, cancer, parasitose digestive], angiodysplasie ou maladie de Rendu-Osler, saignements provoqués [lasthénie de Farjol]) ;
- carence d’apports : grossesses rapprochées, anorexie mentale ;
- malabsorption : maladie cœliaque, résection intestinale importante, gastrite, circonstances altérant l’absorption (consommation excessive de thé), chirurgie bariatrique.
Traitements
Le traitement d’une anémie par carence maritale est avant tout celui de l’étiologie de la carence.
Par ailleurs, une supplémentation martiale peut être instaurée :
- soit par voie orale : de 100 mg à 200 mg/j de sels ferreux (Tardyferon, Fumafer) si possible à jeun (en cas de mauvaise tolérance, cette supplémentation peut être prise lors des repas). Les patients doivent être informés des possibles effets indésirables : coloration des selles en noir, troubles digestifs (douleurs abdominales, diarrhées ou constipation), céphalées (rare). La durée de traitement recommandée est de 4 mois (jusqu’à normalisation de la ferritine) ;
- soit par voie parentérale : elle est indiquée en cas de résistance au traitement oral (traitement intraveineux [Venofer]). Des réactions d’anaphylaxie sont décrites lors de la perfusion. Par ailleurs, d’autres effets indésirables ont été notés, comme des céphalées, nausées, vomissements.
Anémie inflammatoire
Une inflammation chronique peut entraîner une anémie microcytaire par impossibilité de synthèse de l’hème par séquestration du fer par les macrophages, inhibition de l’érythropoïèse et diminution de la synthèse d’EPO.
Bilan biologique
Au niveau biologique, la présence d’un syndrome inflammatoire se traduit par une augmentation de la CRP, de l’haptoglobine, des α-2-globulines et du fibrinogène.
Bilan étiologique
Les étiologies des syndromes inflammatoires chroniques sont nombreuses, et l’orientation diagnostique doit tenir compte du contexte clinique (interrogatoire et examen clinique).
Parmi les étiologies les plus fréquentes, on retrouve les maladies de système telles que le lupus ou la maladie de Horton, les néoplasies et les infections.
Traitement
Le traitement est celui de l’étiologie.
Autres causes d’anémie microcytaire
Dans le cas d’anémie microcytaire sans carence martiale et sans syndrome inflammatoire, le bilan peut être complété à la recherche :
- d’une thalassémie (électrophorèse de l’hémoglobine) ;
- d’une anémie sidéroblastique (myélogramme) ;
- d’un saturnisme (plombémie, plomburie) ;
- d’une hyperthyroïdie (TSH, T4L) ;
- d’une carence en vitamines B1 et B6 (dosages des vitamines B1 et B6) ;
- d’un déficit en cuivre.
Anémies normomacrocytaires arégénératives
La plupart des anémies normomacrocytaires arégénératives sont d’origine centrale. En revanche, avant de réaliser une analyse médullaire, il faut éliminer quelques étiologies telles que :
- l’insuffisance rénale chronique (normocytaire) ;
- l’hypothyroïdie (macrocytaire) ;
- l’anémie inflammatoire débutante (normocytaire).
Une fois ces causes éliminées, une analyse médullaire permet de faire la différence entre les étiologies à moelle riche et celles à moelle pauvre.
Anémies à moelle riche
Carence en vitamines B9 et B12
Les vitamines B12 (cobalamines) et B9 (folates) sont des précurseurs nucléotidiques indispensables à la synthèse de l’ADN. L’absence de ces vitamines ralentit les mitoses au cours de l’érythropoïèse.
Clinique
Comme lors de l’anémie par carence martiale, on retrouve un syndrome anémique d’installation progressive et habituellement bien toléré. Par ailleurs, on retrouve un syndrome carentiel. Dans le contexte des carences vitaminiques B9-B12, le syndrome carentiel est dominé par des atrophies des muqueuses (glossite de Hunter).
Par ailleurs, il existe une caractéristique clinique rare propre à la carence en vitamine B12 : la sclérose combinée de la moelle (syndrome cordonal postérieur et syndrome pyramidal).
Démarche diagnostique
L’hémogramme retrouve une anémie mégaloblastique (VGM > 120 fl) arégénérative. Le seul diagnostic à évoquer face à une anémie macrocytaire avec un VGM > 120 fl est celui de carence en vitamines B9 et/ou B12. Une leuconeutropénie et une thrombopénie peuvent être associées. On peut retrouver des stigmates d’hémolyse liés à l’hémolyse intramédullaire.
Le frottis retrouve une atteinte de toutes les lignées (neutrophiles hypersegmentés, plaquettes géantes, anisocytose).
Le myélogramme retrouve une moelle riche, « bleue », car les cellules sont de grande taille, avec asynchronisme de maturation nucléocytoplasmique. Le myélogramme est recommandé pour ne pas passer à côté d’autres causes d’anémie arégénérative qui pourraient être associées à des carences, notamment les hémopathies.
Par ailleurs, il faut réaliser les dosages vitaminiques B9 et B12.
Bilan étiologique
Après affirmation de la carence vitaminique, l’enjeu est d’en définir l’étiologie.
Il existe 4 grands mécanismes de carence en vitamine B9 :
- carence d’apports : anorexie mentale, alcoolisme, dénutrition ;
- majoration des besoins : grossesses rapprochées, anémie hémolytique chronique ;
- malabsorption : maladie cœliaque, maladie de Crohn, insuffisance pancréatique, résection jéjunale importante ;
- iatrogénie : Bactrim, méthotrexate.
Il existe 2 grands mécanismes de carence en vitamine B12 :
- carence d’apports : anorexie mentale, régime végétalien strict ;
- malabsorption : anémie de Biermer (v. infra), gastrite atrophique, maladie de Crohn, maladie cœliaque, gastrectomie totale, résection iléale.
Traitement
Le traitement d’une anémie par carence vitaminique est avant tout celui de l’étiologie de la carence.
Une supplémentation en vitamine B9 peut être assurée par une voie orale (Spéciafoldine 5 mg par jour pendant 1 mois). Afin de supplémenter une carence en vitamine B12, un traitement d’attaque par traitement intramusculaire 1 000 µg/j pendant 10 jours puis 1 000 µg/mois à vie. Il a été cependant démontré qu’une supplémentation peut se faire par voie orale uniquement.
Maladie de Biermer
La maladie de Biermer est la principale cause de carence en vitamine B12. Il s’agit d’une maladie auto-immune responsable d’une atrophie de la muqueuse gastrique caractérisée par la présence d’auto-anticorps antifacteur intrinsèque et anticorps anticellules pariétales.
Le diagnostic est confirmé par la présence d’une carence en vitamine B12 et des auto-anticorps associés. Les explorations digestives hautes permettent d’identifier une gastrite atrophique. Le traitement repose sur l’administration de vitamine B12 par voie parentérale.
La recherche d’autre pathologie auto-immune est indispensable dans ce contexte.
Myélodysplasie
Les syndromes myélodysplasiques sont des pathologies clonales des cellules souches myéloïdes associées à des anomalies morphologiques qualitatives des lignées myéloïdes.
Envahissement médullaire
Il peut être secondaire à un envahissement de lymphopathie (lymphome agressif ou indolent) ou de métastase de cancer solide. Les leucémies aiguës (prolifération blastique) entraînent aussi une anémie à moelle riche.
Anémie à moelle pauvre
Syndrome myéloprolifératif : myélofibrose (ou splénomégalie myéloïde)
Il s’agit d’un syndrome myéloprolifératif caractérisé par une fibrose médullaire extensive. Elle peut être primitive ou secondaire à d’autres pathologies myéloprolifératives (polyglobulie de Vaquez, thrombocytopénie essentielle).
Aplasie médullaire
L’aplasie médullaire est une insuffisance médullaire quantitative définie par l’appauvrissement du tissu myéloïde sans anomalie qualitative des cellules. Les étiologies sont diverses : congénitales (maladie de Fanconi), acquises (hémoglobinurie paroxystique nocturne), iatrogènes, infectieuses, idiopathiques.
Érythrobastopénie
L’érythroblastopénie est une atteinte spécifique de la lignée érythrocytaire. Les étiologies sont extrêmement variables : iatrogénie, auto-immunité, congénitale (maladie de Blackfan-Diamond), infectieuse (parvovirus B19), idiopathique.
Anémies normomacrocytaires régénératives
Définition
Le caractère régénératif des anémies normocytaires signe leur origine périphérique. Il ne s’agit pas d’un défaut de production d’érythrocytes mais d’une destruction anormale. La macrocytose est liée au raccourcissement de l’érythropoïèse.
Hémolyse
L’hémolyse est associée à une triade biologique :
- augmentation des LDH (marqueur de lyse cellulaire aspécifique) ;
- augmentation de la bilirubine libre (l’hème est dégradée par l’hème oxydase pour produire la biliverdine puis la bilirubine) ;
- effondrement de l’haptoglobine (consommation pour capter l’hémoglobine libre dans le sang).
La présence de schizocytes (fragments de globules rouges) sur le frottis sanguin signe le caractère mécanique de l’anémie.
Au niveau clinique, on distingue l’hémolyse intravasculaire et intratissulaire.
Lors des hémolyses intravasculaires, les érythrocytes sont détruits directement dans les vaisseaux par l’action du complément. Il s’agit d’un tableau aigu, associant de la fièvre, une hypotension pouvant être associée à des signes de choc, des troubles digestifs (nausées, diarrhées), des douleurs lombaires ou abdominales. On retrouve des urines dites « porto », secondaires à l’hémoglobinurie, un ictère et une possible insuffisance rénale.
Lors des hémolyses intratissulaires, les érythrocytes sont détruits dans les tissus (phagocytose). Cela entraîne un tableau moins bruyant, chronique. On retrouve fréquemment une splénomégalie.
Les anémies hémolytiques sont réparties en fonction de la nature corpusculaire (hémolyse liée à une anomalie du globule rouge lui-même, anomalies le plus souvent congénitales) ou extracorpusculaire (hémolyse liée à un agent extérieur, anomalies acquises).
Anémie hémolytique extracorpusculaire
Anémie hémolytique auto-immune
Les anémies hémolytiques auto-immunes (AHAI) sont secondaires à la fixation d’auto-anticorps sur les hématies. Il s’agit principalement d’une hémolyse intravasculaire.
Le diagnostic est assuré par le test de Coombs direct (test direct à l’antiglobuline). Il permet de détecter des anticorps fixés sur les hématies. Selon la température de fixation des anticorps (37 °C ou 4 °C), on distingue les AHAI à anticorps chauds ou à anticorps froids dont les étiologies sont différentes (
La prise en charge d’une AHAI est souvent une urgence et nécessite un avis spécialisé. Le traitement de l’étiologie reste la priorité. Une transfusion de culots érythrocytaires peut être réalisée en cas de mauvaise tolérance de l’anémie. Par ailleurs, une supplémentation en acide folique est indispensable pour aider la régénération médullaire.
Microangiopathie thrombotique
Ce syndrome est une urgence absolue. Il s’agit d’une activation plaquettaire anormale entraînant la formation de microthrombi diffus (thrombopénie de consommation). En obstruant la lumière endothéliale, les microthrombi entraînent des turbulences du flux sanguin et une hémolyse mécanique des hématies.
Le bilan biologique retrouve une anémie hémolytique mécanique (présence de schizocytes sur le frottis) et une thrombopénie, et parfois une insuffisance rénale. Au niveau clinique, les patients peuvent présenter de la fièvre ou des troubles neurologiques.
Une fois le diagnostic de microangiopathie thrombotique retenu, la recherche de l’étiologie doit être effectuée rapidement : infections (notamment, chez l’enfant, Escherichia coli O157:H7), iatrogénie, maladie de système, cancers, hypertension artérielle maligne, déficit en ADAMTS-13 (congénital ou acquis), HELLP syndrome chez la femme enceinte.
La prise en charge (notamment étiologique) doit être rapide. Il faut au maximum éviter la transfusion de plaquettes, qui pourrait majorer la formation de microthrombi et donc l’hémolyse.
Hémolyse corpusculaire par anomalie de membrane
Maladie de Minkowski-Chauffard, ou sphérocytose héréditaire
Il s’agit d’une anémie hémolytique corpusculaire chronique secondaire à une anomalie d’une protéine du cytosquelette sous-membranaire des érythrocytes. Cela entraîne une déformation des globules rouges sous forme de sphère (sphérocytes). Ces sphérocytes sont peu déformables et sont séquestrés puis détruits dans la rate (hémolyse intratissulaire).
Différentes mutations sont responsables de cette anomalie, certaines sont de transmission autosomique dominante (deux tiers des cas) et d’autres sont de transmission autosomique récessive (un tiers des cas).
Le diagnostic est souvent réalisé dans l’enfance. On retrouve une hémolyse chronique (syndrome anémique modéré, subictère, splénomégalie) mais la maladie évolue aussi par crises d’hyperhémolyse (syndrome anémique important et ictère franc) souvent révélées après un facteur déclenchant (infections, prise médicamenteuse).
Le diagnostic est réalisé grâce à la confirmation de l’hémolyse, la présence de sphérocytes sur le frottis sanguin et la réalisation d’une ektacytométrie (quantification de la déformation des hématies).
Cette pathologie peut se compliquer d’érythroblastopénie en cas d’infection par le parvovirus B19 et de lithiase vésiculaire pigmentaire, avec risque de colique hépatique ou de cholécystite aiguë.
La réalisation d’une splénectomie constitue le traitement de référence (après vaccinations et sous couvert de traitement antibiotique préventif au long cours). Par ailleurs, la supplémentation en acide folique est indispensable afin d’aider à la régénération médullaire. De même, une enquête familiale est indispensable.
Hémoglobinurie paroxystique nocturne
Il s’agit de la seule anémie hémolytique corpusculaire acquise. Elle est due à une mutation du gène PIG-A régulant la synthèse de la protéine d’ancrage GPI sur les cellules hématopoïétiques. Le GPI permet l’ancrage sur la surface des cellules circulantes des protéines CD55 et CD59 dont le rôle est l’inhibition du complexe lytique du complément. En l’absence de GPI et donc de CD55 et CD59, le complément reste activé et entraîne la lyse des hématies.
Le tableau clinique est dominé par une hémolyse intravasculaire paroxystique (volontiers nocturne) associant un syndrome anémique, une hémoglobinurie, un ictère cutanéo-muqueux et parfois des douleurs lombaires. On peut fréquemment retrouver un facteur déclenchant aux crises d’hémolyse : infections, vaccination, chirurgie ou effort physique intense.
Par ailleurs, les patientes peuvent présenter des thromboses veineuses (principalement dans le réseau profond).
Le diagnostic de certitude est établi avec la réalisation d’un immunophénotypage sanguin : détection d’une population déficitaire en CD55 et CD59). L’hémoglobinurie paroxystique nocturne peut se compliquer d’aplasie médullaire.
Le traitement curatif reste l’allogreffe, mais il existe aussi un traitement symptomatique (anticorps monoclonal anti-fraction C5 du complément).
Hémolyse corpusculaire enzymatique
Déficit en G6PD
Cette pathologie touche principalement les populations du pourtour méditerranéen, d’Afrique noire et d’Asie. Étant de transmission liée à l’X, il existe une forte prédominance masculine.
Il s’agit d’une anémie hémolytique corpusculaire aiguë par accumulation de radicaux libres dans les hématies secondaires au déficit en glucose-6-phosphate déshydrogénase (G6PD). Cette accumulation de stress oxydatif est responsable de la fragilité des membranes, et donc de l’hémolyse. Tout facteur responsable d’une augmentation des radicaux libres (notamment les médicaments) est susceptible d’entraîner une crise d’hémolyse. C’est pourquoi il est primordial de rechercher un facteur déclenchant : iatrogène (sulfamides, antipaludéens, quinolones, vitamine K, dérivés nitrés), consommation de fèves, infections virales (hépatites) et bactériennes.
Biologiquement, on retrouve une anémie hémolytique, et sur le frottis sanguin des corps de Heinz. Hors période d’hémolyse, on peut réaliser un dosage de G6PD. Le traitement est avant tout l’éviction des facteurs déclenchants et donc une éducation stricte des patients.
Déficit en pyruvate kinase
Le déficit en pyruvate kinase est de transmission autosomique récessive. Cette enzyme sert à la synthèse d’ATP. En son absence, la membrane des hématies est instable, et cela entraîne une hémolyse.
Le tableau clinique est celui d’une hémolyse chronique. Le frottis sanguin retrouve des hématies crénelées, et le dosage de pyruvate kinase est effondré.
Dans les formes sévères, la splénectomie peut être discutée. Comme dans toutes les hémolyses chroniques, il est important de penser à la supplémentation en acide folique.
Anémies hémolytiques corpusculaires par anomalie de l’hémoglobine
Généralités
L’hémoglobine (Hb) est composée de 4 molécules de globines identiques 2 à 2. La sous-unité α (alpha) est toujours présente quel que soit le type d’Hb. Chez le fœtus, l’HbF (α2γ2) est prédominante alors que chez l’enfant et l’adulte il s’agit de l’HbA (α2β2).
Les hémoglobinopathies sont responsables d’anémie.
Drépanocytose
Cette pathologie de transmission autosomique récessive (les patients hétérozygotes ne sont pas symptomatiques) touche principalement les populations d’Afrique, du Moyen-Orient et du pourtour méditerranéen.
Il s’agit d’une hémoglobinopathie secondaire à une mutation ponctuelle au niveau du gène de la globine β (cette globine anormale est alors appelée HbS). Cette anomalie va entraîner un changement de la structure de l’hémoglobine et donc des hématies qui prendront alors une forme de faucille (falciforme). Cette anomalie structurelle réduit la déformabilité des hématies, ce qui entraîne des agrégats dans la lumière des vaisseaux (> thrombose). Ces hématies bloquées sont détruites par le système macrophagique (hémolyse).
Le bilan biologique retrouve une anémie normocytaire régénérative, la présence de drépanocytes sur le frottis sanguin. Sur l’électrophorèse de l’hémoglobine, il n’existe pas d’hémoglobine adulte A adulte ; en revanche, on note la présence d’une hémoglobine S importante (normalement absente chez les sujets sains). Enfin, il existe une HbF (fœtale) anormalement élevée.
Les manifestations cliniques apparaissent après les 2-3 premiers mois de vie (parallèlement à l’apparition de la chaîne β de globine). En dehors des épisodes d’hémolyse aigus, le tableau clinique retrouve une splénomégalie, un subictère et un syndrome anémique modéré. Lors d’un facteur déclenchant (fièvre, infection, déshydratation, hypoxie), les patients peuvent présenter des crises vaso-occlusives qui se manifestent par des douleurs intenses et brutales de localisation diverse (ostéoarticulaire, thoracique, abdominale). Lors de ces accidents vaso-occlusifs, il faut rechercher en urgence des signes de gravité : accident vasculaire cérébral, priapisme, syndrome thoracique aigu (hypoxie, douleurs thoraciques, anomalies radiologiques), occlusion de la veine centrale de la rétine. Le traitement des crises vaso-occlusives nécessite en priorité la prise en charge du facteur déclenchant, puis la mise en place d’antalgiques (souvent de palier 3), et une oxygénothérapie systématique. En cas de fièvre, il faut débuter immédiatement une antibiothérapie (afin de couvrir une potentielle infection par le pneumocoque). En cas d’inefficacité des mesures classiques de prise en charge, il est possible de réaliser une exsanguinotransfusion afin de faire diminuer le taux d’HbS.
Les complications chroniques de la drépanocytose sont nombreuses (secondaires aux crises vaso-occlusives répétées) : retard staturopondéral, retard pubertaire, séquelles neurologiques (post-AVC), oculaires (rétinopathie proliférante, avec risque d’hémorragie ou de décollement rétinien et de cécité), cardiovasculaires (hypertension artérielle pulmonaire [HTAP], cardiomyopathie dilatée), pulmonaires, néphrologiques (néphropathie glomérulaire), hépatobiliaires (sur lithiase), ostéo-articulaires (nécrose de la tête fémorale). Des examens complémentaires doivent donc être réalisés régulièrement pour assurer la surveillance de ces complications.
Par ailleurs, du fait des infarctus spléniques répétés, les patients drépanocytaires développent une asplénie fonctionnelle et donc une susceptibilité aux infections (notamment aux germes encapsulés). Il est donc indispensable de maintenir les vaccinations à jour et d’éduquer les patients au risque infectieux.
Thalassémie
Bien qu’étant une anémie hémolytique corpusculaire par anomalie de l’hémoglobine, l’anémie secondaire à la thalassémie est en réalité une anémie microcytaire. Elle est de transmission autosomique récessive.
Les thalassémies majeures peuvent se compliquer de déglobulisation aiguë, notamment après une infection. Les transfusions sont la base de la prise en charge et peuvent entraîner une hémochromatose secondaire, qu’il faut prendre en charge par des chélateurs du fer afin d’éviter les complications surajoutées.
Alphathalassémies
Il existe 4 gènes (2 par chromosome) codant pour la sous-unitéαde la globine. Il existe donc 4 formes cliniques en fonction du nombre de gène α délétés ou mutés :
- 1 gène délété ou muté : électrophorèse de l’hémoglobine normale, microcytose isolée, pas de répercussion clinique ;
- 2 gènes délétés ou mutés : α ; thalassémie mineure : électrophorèse de l’hémoglobine quasi normale (faible diminution de l’HbA2 : α2γ2), microcytose isolée, pas de répercussion clinique ;
- 3 gènes délétés ou mutés : α-thalassémie majeure ou hémoglobinose H : les sous-unités α sont en parties remplacées par des sous-unités β (HbH = β4), anémie microcytaire hypochrome profonde ;
- 4 gènes délétés ou mutés : anasarque de Bart. Les sous-unités α sont remplacées par des sous-unités γ (Hb Bart : γ4). Ceci entraîne une thalassémie non viable avec mort fœtale in utero.
Bêtahalassémies
Il existe deux gènes codant pour la sous-unité β de la globine (1 sur chaque chromosome).
Il existe donc 2 formes cliniques en fonction du nombre de gène β délétés ou mutés :
- 1 gène muté ou délété. β-thalassémie mineure : électrophorèse de l’hémoglobine quasi normale (augmentation modérée de l’HbA2), anémie microcytaire modérée, pas de répercussion clinique ;
- 2 gènes mutés ou délétés. β-thalassémie majeure (anémie de Cooley) : électrophorèse de l’hémoglobine : absence totale d’HbA, replacée par HbF. Anémie microcytaire profonde, splénomégalie par hémolyse.
Autres anémies hémolytiques
Iatrogène
Certains médicaments entraînent des anémies hémolytiques.
Il existe 2 mécanismes distincts :
- le médicament vient se fixer sur les hématies, et l’anticorps dirigé contre le médicament est responsable de l’hémolyse ;
- le complexe anticorps-médicaments vient s’absorber sur les hématies, qui sont alors détruites par l’action du complément.
Hémolyse cardiaque
Il existe un phénomène d’hémolyse chez les patients porteurs d’une valve mécanique ou calcifiée.
Régénération
À la suite d’une hémorragie en sortie d’aplasie post-chimiothérapie, les patients présentent une anémie normocytaire régénérative sans hémolyse.
Conclusion
L’anémie est un symptôme fréquent pouvant avoir des causes extrêmement variables. Il est primordial de bien savoir définir les caractéristiques de l’anémie afin d’orienter plus facilement les examens complémentaires à la recherche de son étiologie et donc permettre un traitement adapté (
POINTS FORTS À RETENIR
L’anémie est une pathologie fréquente définie par une baisse de l’hémoglobine en dessous de seuils qui sont variables selon l’âge et le sexe.
L’identification des caractéristiques de l’anémie (analyse des VGM, TCMH et taux de réticulocytes) est une étape cruciale pour la compréhension et la démarche diagnostique.
La tolérance de l’anémie est inversement liée à la rapidité d’installation.
Les carences en vitamine B9 et B12 entraînent des anémies mégaloblastiques (VGM > 120 fl).
Devant une anémie normomacrocytaire arégénérative, et une fois avoir éliminé une insuffisance rénale chronique, une hypothyroïdie et une anémie inflammatoire débutante, il faut systématiquement compléter le bilan par un myélogramme.
L’association d’une anémie normomacrocytaire hémolytique et d’une thrombopénie doit faire rechercher en urgence une microangiopathie thrombotique.
Ne pas oublier le bilan prétransfusionnel.
Les anémies normomacrocytaires régénératives sont dominées par les cas d’hémolyse.
Anémie chez l’adulte et l’enfant
L’anémie de l’adulte est une question récurrente aux examens. En effet, les étiologies variables des anémies permettent d’en faire une question pivot pour beaucoup de dossiers transversaux. L’axe principal sera bien évidemment le bilan diagnostique et la hiérarchisation des examens complémentaires en fonction des caractéristiques initiales de l’anémie. Ensuite, le bilan étiologique permet de créer des dossiers très différents. Par exemple, une anémie ferriprive peut faire diagnostiquer un cancer colique, un cancer de l’utérus ou un ulcère gastroduodénal. Une anémie inflammatoire peut être le symptôme initial d’une maladie de système, comme une maladie de Horton ou un lupus érythémateux disséminée. Par ailleurs, un dossier d’anémie peut être l’occasion de poser des questions sur les modalités et possibles complications des transfusions. Enfin, il est indispensable de connaître les modalités des traitements en cas de carence martiale, en B12 ou en folates.
 Encadrés
Encadrés