Les anémies hémolytiques auto-immunes sont caractérisées par une hémolyse, un caractère régénératif (dans 90 % des cas) et un test direct à l’antiglobuline (TDA) positif dans 95 % des cas. On en distingue deux formes principales : celles à anticorps chauds (TDA positif en IgG) et celles à anticorps froids (TDA négatif en IgG mais positif en C3d), qui diffèrent par leurs causes sous-jacentes et par leur traitement.
Les hématies sont produites dans la moelle osseuse à partir des cellules souches myéloïdes différenciées en érythroblastes. L’érythropoïèse aboutit à la synthèse des réticulocytes (jeunes globules rouges). Le volume moyen des réticulocytes est de 112 fL* chez le sujet sain (133 fL en cas d’anémie hémolytique auto-immune [AHAI]), contre 90 fL pour les hématies, expliquant l’augmentation du volume globulaire moyen observée dans la plupart des anémies régénératives.1 Les globules rouges ont une durée de vie moyenne de 120 jours avant d’être détruits dans la pulpe rouge de la rate.
L’hémolyse se définit par une diminution de cette durée de vie, liée à la destruction des hématies qui entraîne une libération plasmatique des dérivés de l’hémoglobine. Cette hémolyse est due le plus souvent à une destruction accrue des globules rouges dans le secteur intravasculaire (hémolyse intravasculaire) et/ou dans le foie ou la rate (hémolyse intratissulaire). Dans de rares cas, un tableau biologique mimant celui d’une anémie d’allure hémolytique peut s’observer en cas d’avortement des progéniteurs érythroïdes dans la moelle osseuse (hémolyse intra-médullaire).
L’hémolyse intravasculaire provoque une hémoglobinémie et une hémoglobinurie, entraînant une coloration brune des urines (urines « rouge porto »). Elle se voit notamment au cours du paludisme, de l’hémoglobinurie paroxystique nocturne, des hémolyses mécaniques, du déficit en G6PD et à un degré moindre au cours de la maladie des agglutinines froides.
L’hémolyse intratissulaire peut être la conséquence d’une perte de la déformabilité du globule rouge entraînant sa destruction par la pulpe rouge de la rate (anomalies de l’hémoglobine, anomalie de membrane) ou de la présence d’auto-anticorps à la surface du globule rouge (AHAI à anticorps chauds).
L’hémolyse intramédullaire se caractérise par des taux souvent très élevés de lacticodéhydrogénase (LDH), la présence possible de schizocytes (tableau de « pseudomicro-angiopathie thrombotique ») mais, en revanche, par un taux de réticulocytes normal qui tranche avec la macrocytose. Elle s’observe dans des carences profondes en vitamine B12 et plus rarement en vitamine B9.
L’hémolyse peut avoir plusieurs conséquences :
– une anémie (si la production médullaire ne compense pas la destruction accrue des hématies) ;
– une splénomégalie ;
– un risque accru de thrombose ;2, 3
– une carence en acide folique (nécessitant une supplémentation systématique).
L’hémolyse se définit par une diminution de cette durée de vie, liée à la destruction des hématies qui entraîne une libération plasmatique des dérivés de l’hémoglobine. Cette hémolyse est due le plus souvent à une destruction accrue des globules rouges dans le secteur intravasculaire (hémolyse intravasculaire) et/ou dans le foie ou la rate (hémolyse intratissulaire). Dans de rares cas, un tableau biologique mimant celui d’une anémie d’allure hémolytique peut s’observer en cas d’avortement des progéniteurs érythroïdes dans la moelle osseuse (hémolyse intra-médullaire).
L’hémolyse intravasculaire provoque une hémoglobinémie et une hémoglobinurie, entraînant une coloration brune des urines (urines « rouge porto »). Elle se voit notamment au cours du paludisme, de l’hémoglobinurie paroxystique nocturne, des hémolyses mécaniques, du déficit en G6PD et à un degré moindre au cours de la maladie des agglutinines froides.
L’hémolyse intratissulaire peut être la conséquence d’une perte de la déformabilité du globule rouge entraînant sa destruction par la pulpe rouge de la rate (anomalies de l’hémoglobine, anomalie de membrane) ou de la présence d’auto-anticorps à la surface du globule rouge (AHAI à anticorps chauds).
L’hémolyse intramédullaire se caractérise par des taux souvent très élevés de lacticodéhydrogénase (LDH), la présence possible de schizocytes (tableau de « pseudomicro-angiopathie thrombotique ») mais, en revanche, par un taux de réticulocytes normal qui tranche avec la macrocytose. Elle s’observe dans des carences profondes en vitamine B12 et plus rarement en vitamine B9.
L’hémolyse peut avoir plusieurs conséquences :
– une anémie (si la production médullaire ne compense pas la destruction accrue des hématies) ;
– une splénomégalie ;
– un risque accru de thrombose ;2, 3
– une carence en acide folique (nécessitant une supplémentation systématique).
Quelle démarche diagnostique ?
Devant une anémie, le diagnostic d’anémie hémolytique auto-immune se déroule en plusieurs étapes.
Analyse du Volume Globulaire Moyen
Le volume globulaire moyen (VGM) est augmenté dans la majorité des anémies hémolytiques auto-immunes, du fait de la réticulocytose.
Recherche du caractère régénératif de l’anémie
Une anémie régénérative (réticulocytes > 150 000/mm3) oriente vers une hémolyse, une hémorragie aiguë récente (extériorisée ou non) en voie de « réparation », ou la correction d’une cause d’anémie arégénérative (supplémentation martiale ou en vitamine B9 ou B12, disparition d’un syndrome inflammatoire, sortie d’aplasie post-chimiothérapie…). On note cependant que 10 à 20 % des anémies hémolytiques auto-immunes sont arégénératives (lorsque l’auto-anticorps cible aussi les réticulocytes ou encore en cas de carence en folates surajoutée, et également au tout début du processus d’hémolyse avant la mise en route de la compensation médullaire).
Recherche du caractère hémolytique de l’anémie
Le diagnostic d’hémolyse repose sur un faisceau d’arguments, LDH augmentée, haptoglobine abaissée, bilirubine libre augmentée, mais aucun de ces trois paramètres n’est spécifique d’une hémolyse. L’élévation de la bilirubine libre semble la plus spécifique (si l’on exclut la maladie de Gilbert), mais sa sensibilité ne dépasse pas 87 %.4 En l’absence de syndrome inflammatoire associé, la baisse du taux d’haptoglobine reste le marqueur le plus sensible d’hémolyse.
Quelle cause ?
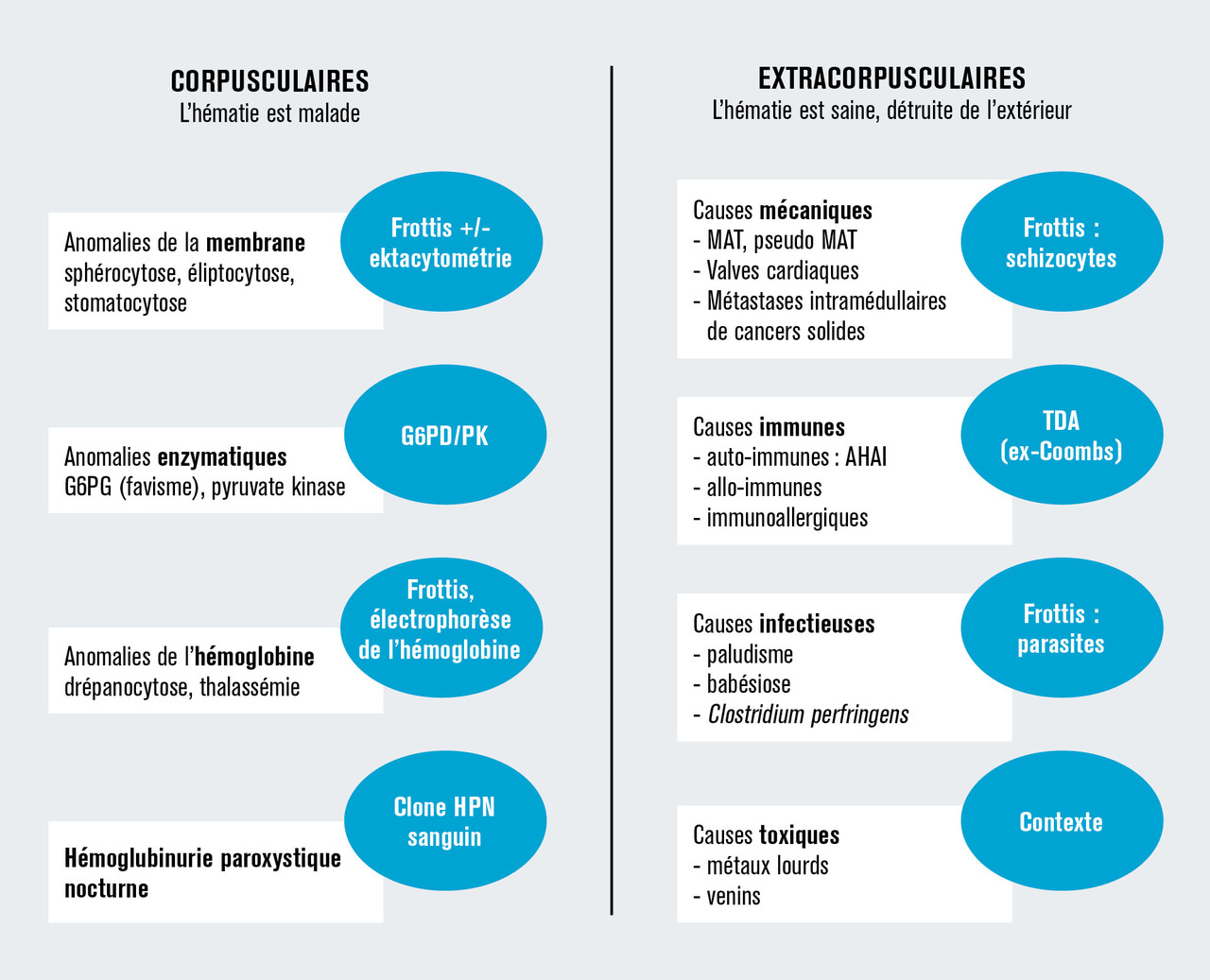
Une fois l’hémolyse retenue, de nombreuses causes congénitales ou acquises sont à envisager (fig. 1 ). Les tests de première intention sont le frottis sanguin et le test direct à l’antiglobuline (TDA) [anciennement test de Coombs direct].
Le frottis sanguin peut montrer des anomalies morphologiques des hématies (anomalies de membrane, anomalie de l’hémoglobine, schizocytose dans les anémies mécaniques) ou la présence de microbes intra-érythrocytaire (paludisme, babésiose). La présence de sphérocytes n’est pas spécifique de la microsphérocytose, elle est classique dans l’anémie hémolytique auto-immune à anticorps chauds, où elle s’observe dans environ 40 % des cas.
Un TDA positif de type Ig et/ou complément (v. infra) permet de confirmer, en l’absence d’autre cause manifeste d’hémolyse, le mécanisme immunologique de l’anémie. Lorsque le patient n’a pas été exposé préalablement à des transfusions, la présence d’IgG et ou de C3 à la surface des hématies attesté par le TDA permet de conclure à un mécanisme auto-immun.
Un TDA négatif ne permet cependant pas d’éliminer une anémie hémolytique auto-immune, 5 % des anémies hémolytiques auto-immunes se présentant avec un TDA négatif.
Une fois le diagnostic d’anémie hémolytique auto-immune posé, il faut répondre à deux questions :
– s’agit-il d’une anémie hémolytique auto-immune à anticorps « chauds » ou « froids » ? Cela dépend de l’optimum thermique de l’anticorps (= la température à laquelle l’auto-anticorps exerce son activité hémolytique maximale) : l’optimum thermique est proche de 37 °C dans les anémies hémolytiques auto-immunes à anticorps chauds. Il est inférieur à 20 °C (le plus souvent entre 0 et 4 °C) en cas d’anticorps froids, appelés agglutinines froides en raison de leur capacité à faire agglutiner les hématies.
En pratique, le TDA fait à température ambiante (20 °C) permet de répondre à cette question (v. infra) ;
– y a-t-il une cause sous-jacente à cette anémie hémolytique auto-immune ?
Ces deux questions sont intimement liées puisque les causes diffèrent avec le caractère chaud ou froid. Le traitement est lui aussi différent.
Le frottis sanguin peut montrer des anomalies morphologiques des hématies (anomalies de membrane, anomalie de l’hémoglobine, schizocytose dans les anémies mécaniques) ou la présence de microbes intra-érythrocytaire (paludisme, babésiose). La présence de sphérocytes n’est pas spécifique de la microsphérocytose, elle est classique dans l’anémie hémolytique auto-immune à anticorps chauds, où elle s’observe dans environ 40 % des cas.
Un TDA positif de type Ig et/ou complément (v. infra) permet de confirmer, en l’absence d’autre cause manifeste d’hémolyse, le mécanisme immunologique de l’anémie. Lorsque le patient n’a pas été exposé préalablement à des transfusions, la présence d’IgG et ou de C3 à la surface des hématies attesté par le TDA permet de conclure à un mécanisme auto-immun.
Un TDA négatif ne permet cependant pas d’éliminer une anémie hémolytique auto-immune, 5 % des anémies hémolytiques auto-immunes se présentant avec un TDA négatif.
Une fois le diagnostic d’anémie hémolytique auto-immune posé, il faut répondre à deux questions :
– s’agit-il d’une anémie hémolytique auto-immune à anticorps « chauds » ou « froids » ? Cela dépend de l’optimum thermique de l’anticorps (= la température à laquelle l’auto-anticorps exerce son activité hémolytique maximale) : l’optimum thermique est proche de 37 °C dans les anémies hémolytiques auto-immunes à anticorps chauds. Il est inférieur à 20 °C (le plus souvent entre 0 et 4 °C) en cas d’anticorps froids, appelés agglutinines froides en raison de leur capacité à faire agglutiner les hématies.
En pratique, le TDA fait à température ambiante (20 °C) permet de répondre à cette question (v. infra) ;
– y a-t-il une cause sous-jacente à cette anémie hémolytique auto-immune ?
Ces deux questions sont intimement liées puisque les causes diffèrent avec le caractère chaud ou froid. Le traitement est lui aussi différent.
Tests biologiques utiles pour le diagnostic
Test direct à l’antiglobuline
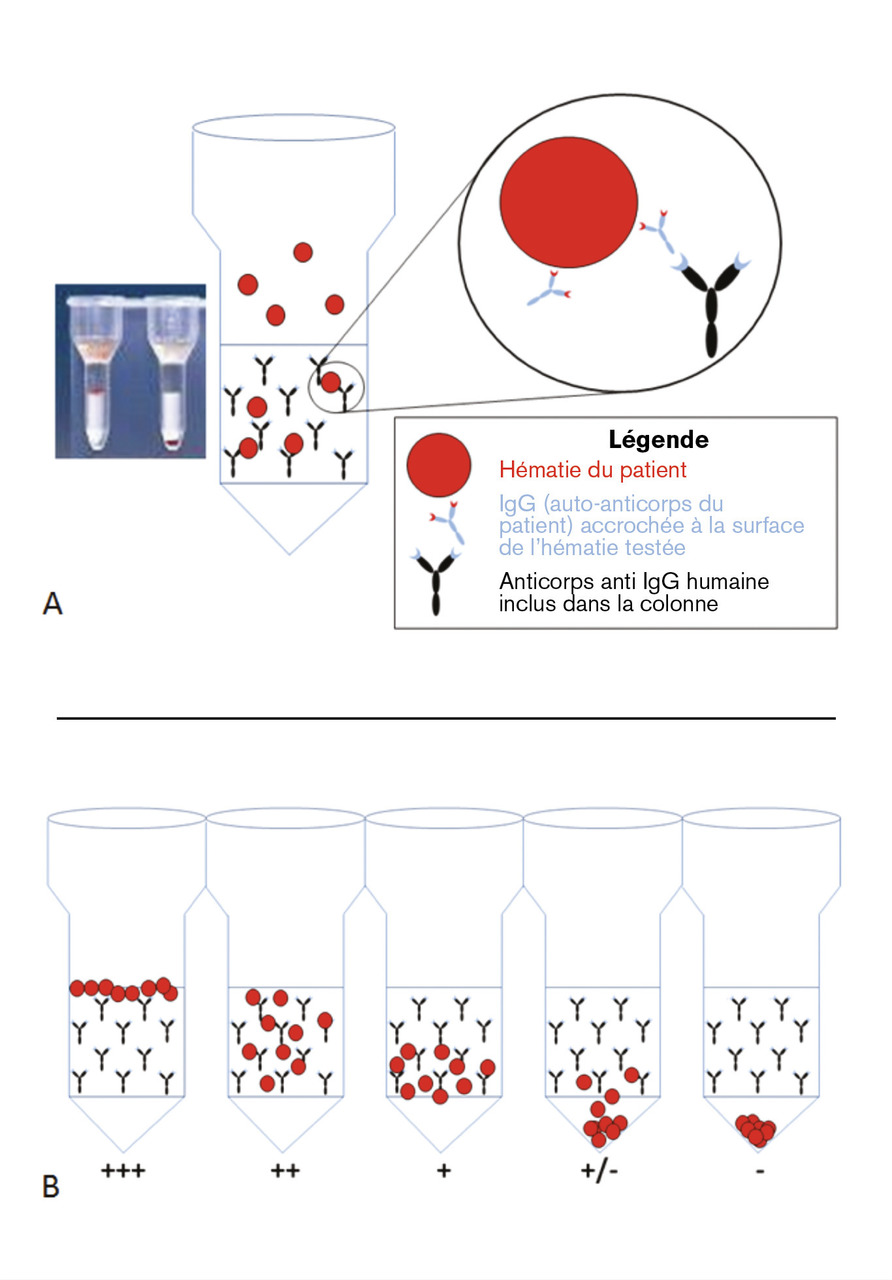
Il recherche la présence d’anticorps et/ou de fractions C3 du complément fixés in vivo à la surface des hématies.
La méthode la plus répandue consiste à séparer les hématies du sérum du patient, puis à les faire migrer dans des colonnes en gel (fig. 2 ) pré-incubées avec une antiglobuline anti-IgG ou un anticorps anti-C3. Si les hématies sont opsonisées par des dépôts de C3 et/ou d’IgG, elles sont piégées dans la colonne, et le test est positif, et inversement. Les résultats sont souvent rendus en semi quantitatif (de 0 à ++++).
La technique de routine n’est réalisée qu’avec l’anti-IgG et l’anti-C3.
Les IgM (responsables des AHAI froides) induisent une forte activation du complément. Leur implication dans l’anémie hémolytique auto-immune est révélée de façon indirecte (C3 très positif, IgG négatif) puisque les IgM ne sont pas recherchées en routine et sont le plus souvent éluées ex vivo du fait de leurs propriétés physicochimiques.
Dans les anémies hémolytiques auto-immunes à anticorps chauds, le TDA est de spécificité IgG ± C3.
Dans les anémies hémolytiques auto-immunes à anticorps froids, le TDA est dans la majorité des cas de spécificité C3 isolée.
Certaines anémies hémolytiques auto-immunes se présentent avec un test de TDA négatif (environ 5 % des cas), soit parce que l’auto-anticorps en cause est une IgA (non recherchée en routine), soit parce que l’anticorps est de trop faible affinité (et donc élué in vitro, même s’il est in vivo capable d’induire l’hémolyse), soit parce que le seuil de détection de la technique est trop faible. En cas de suspicion d’anémie hémolytique auto-immune, si le TDA est rendu négatif, il faut demander un TDA dit « élargi » à la recherche d’un anticorps de type IgA ± une élution pour sensibiliser la technique. Si ces tests sont négatifs, le diagnostic d’anémie hémolytique auto-immune à TDA négatif reste un diagnostic d’exclusion et implique notamment d’avoir éliminé les autres causes d’anémie hémolytique constitutionnelle ou acquise.
La méthode la plus répandue consiste à séparer les hématies du sérum du patient, puis à les faire migrer dans des colonnes en gel (
La technique de routine n’est réalisée qu’avec l’anti-IgG et l’anti-C3.
Les IgM (responsables des AHAI froides) induisent une forte activation du complément. Leur implication dans l’anémie hémolytique auto-immune est révélée de façon indirecte (C3 très positif, IgG négatif) puisque les IgM ne sont pas recherchées en routine et sont le plus souvent éluées ex vivo du fait de leurs propriétés physicochimiques.
Dans les anémies hémolytiques auto-immunes à anticorps chauds, le TDA est de spécificité IgG ± C3.
Dans les anémies hémolytiques auto-immunes à anticorps froids, le TDA est dans la majorité des cas de spécificité C3 isolée.
Certaines anémies hémolytiques auto-immunes se présentent avec un test de TDA négatif (environ 5 % des cas), soit parce que l’auto-anticorps en cause est une IgA (non recherchée en routine), soit parce que l’anticorps est de trop faible affinité (et donc élué in vitro, même s’il est in vivo capable d’induire l’hémolyse), soit parce que le seuil de détection de la technique est trop faible. En cas de suspicion d’anémie hémolytique auto-immune, si le TDA est rendu négatif, il faut demander un TDA dit « élargi » à la recherche d’un anticorps de type IgA ± une élution pour sensibiliser la technique. Si ces tests sont négatifs, le diagnostic d’anémie hémolytique auto-immune à TDA négatif reste un diagnostic d’exclusion et implique notamment d’avoir éliminé les autres causes d’anémie hémolytique constitutionnelle ou acquise.
Dosage des agglutinines froides
Il s’agit d’un test indirect à l’antiglobuline (v. infra) pratiqué à 4 °C. En cas de positivité, le test est réeffectué à différentes dilutions. Le seuil de positivité retenu est 1/64.
Un résultat positif à 1/64 signifie que le sérum du patient, dilué 64 fois, contient encore assez d’agglutinines pour provoquer une agglutination d’hématies à 4 °C.
La présence d’agglutinines froides isolément à titre faible peut se voir notamment chez les sujets âgés et n’est pas forcément pathologique surtout si le TDA est négatif par ailleurs.
Un résultat positif à 1/64 signifie que le sérum du patient, dilué 64 fois, contient encore assez d’agglutinines pour provoquer une agglutination d’hématies à 4 °C.
La présence d’agglutinines froides isolément à titre faible peut se voir notamment chez les sujets âgés et n’est pas forcément pathologique surtout si le TDA est négatif par ailleurs.
Certains tests ne sont pas nécessaires en pratique clinique de routine
Test indirect à l’antiglobuline
En dehors de la recherche spécifique et de la titration d’agglutinines froides en présence de C3 détecté à la surface des hématies par le TDA, ce test n’a pas d’intérêt en routine en cas de TDA positif de type IgG isolé. Il a, en revanche, un intérêt chez les patients exposés à des transfusions préalables afin de faire la part entre auto- et allo-anticorps. Le principe repose sur la mise en contact du sérum du patient avec des hématies témoins. Les agglutinines éventuellement présentes dans le sérum testé sont ensuite révélées à la surface des hématies par la technique décrite ci-dessus (technique du TDA).
Cette technique est celle utilisée en routine pour la recherche d’agglutinines irrégulières (RAI), c’est-à-dire d’agglutinines autres que les anti-A et anti-B (du système ABO), en pré- et post-transfusionnel.
Cette technique est celle utilisée en routine pour la recherche d’agglutinines irrégulières (RAI), c’est-à-dire d’agglutinines autres que les anti-A et anti-B (du système ABO), en pré- et post-transfusionnel.
Test d’élution
Ce test permet de « décrocher » les agglutinines fixées sur les hématies (par la chaleur ou l’acidité) et de déterminer la spécificité des anticorps, c’est-à-dire la cible reconnue à la surface du globule rouge. Il peut être utile notamment en cas de TDA négatif avec une forte suspicion d’anémie hémolytique auto-immune ou dans les cas complexes (il permet par exemple de différencier allo- et auto-anticorps) ou encore de suspicion d’anémie hémolytique auto-immune induite par un médicament.
Épidémiologie et classification des anémies hémolytiques auto-immunes
Les anémies hémolytiques auto-immunes sont des maladies rares : leur incidence est estimée à environ 8 à 10 cas/million d’habitants. En France, il y aurait donc environ 600 nouveaux cas par an. Elles peuvent atteindre tous les âges de la vie.
On distingue plusieurs formes cliniques (tableau 1 ). Les anémies hémolytiques auto-immunes à anticorps chauds représentent 70 à 80 % de l’ensemble des anémies hémolytiques auto-immunes chez l’adulte.
On distingue plusieurs formes cliniques (
Anémies hémolytiques auto-immunes à anticorps chauds
Ce sont les formes les plus fréquentes. Elles peuvent survenir à tout âge de la vie mais prédominent après l’âge de 40 ans chez l’adulte, avec une légère surreprésentation féminine (odds ratio : 1-1,5).
Le mode de révélation est clinique dans plus de 80 % des cas (syndrome anémique, urines foncées, ictère…). Plus rarement, il s’agit d’une découverte biologique fortuite.
L’auto-anticorps est le plus souvent une IgG (IgG1 ou IgG3 surtout), détectable par le TDA, qui peut entraîner une activation de la voie classique du complément, ce qui fait que le TDA est également positif avec l’anti-C3 dans environ 50 à 60 % des cas.
Dans environ 50 % des cas l’anémie hémolytique est isolée et alors qualifiée de forme « primaire ». Les formes secondaires sont donc fréquentes et à rechercher systématiquement. Letableau 2 résume les différentes causes et les explorations proposées en première intention par le centre de référence des cytopénies auto-immunes (protocole national de diagnostic et des soins révisé en 2017).
Dans une série descriptive de 60 anémies hémolytiques auto-immunes à anticorps chauds suivie dans le centre de référence français, on notait une mortalité globale de 8 % après une médiane de suivi à 46 mois,4 mais des taux de mortalité allant jusqu’à 17 à 20 % ont été rapportées dans la littérature.5 Les causes de mortalité sont liées aux complications de la maladie (syndromes coronariens, thromboses), à la maladie sous-jacente (hémopathies lymphoïdes) et aux complications des traitements immunosuppresseurs (infections).
Le mode de révélation est clinique dans plus de 80 % des cas (syndrome anémique, urines foncées, ictère…). Plus rarement, il s’agit d’une découverte biologique fortuite.
L’auto-anticorps est le plus souvent une IgG (IgG1 ou IgG3 surtout), détectable par le TDA, qui peut entraîner une activation de la voie classique du complément, ce qui fait que le TDA est également positif avec l’anti-C3 dans environ 50 à 60 % des cas.
Dans environ 50 % des cas l’anémie hémolytique est isolée et alors qualifiée de forme « primaire ». Les formes secondaires sont donc fréquentes et à rechercher systématiquement. Le
Dans une série descriptive de 60 anémies hémolytiques auto-immunes à anticorps chauds suivie dans le centre de référence français, on notait une mortalité globale de 8 % après une médiane de suivi à 46 mois,4 mais des taux de mortalité allant jusqu’à 17 à 20 % ont été rapportées dans la littérature.5 Les causes de mortalité sont liées aux complications de la maladie (syndromes coronariens, thromboses), à la maladie sous-jacente (hémopathies lymphoïdes) et aux complications des traitements immunosuppresseurs (infections).
Traitement des formes primaires
Les cas d’anémie hémolytique auto-immune à anticorps chauds avec hémolyse compensée et un taux d’hémoglobine supérieur à 10 g/dL et stable en l’absence de traitement restent l’exception et dans 95 % des cas un traitement se justifie sachant que les rémissions spontanées sont très rares.
Le traitement de première ligne des formes « primaires » repose sur la corticothérapie à la dose initiale de 1 à 1,5 mg/kg/j à maintenir pendant 3 à 4 semaines suivie d’une décroissance progressive et, si possible, d’un arrêt en 3 à 6 mois. Ce traitement permet de guérir à lui seul environ un tiers des patients.6, 7
Le « dogme » ancien suggérant des doses plus fortes de corticoïdes et sur une durée prolongée (2 mg/kg initialement puis décroissance sur 2 ans) n’a plus de raison d’être car il ne reposait pas sur des études prospectives, et une durée prolongée de la corticothérapie ne diminue pas le taux de corticodépendance.
En deuxième ligne, en cas d’inefficacité primaire de la corticothérapie évaluée à 1 mois (rare) ou plus souvent de corticodépendance à haut niveau ou de rechute, le rituximab est privilégié. En effet, il a démontré son efficacité au travers de deux études prospectives randomisées dont une en double aveugle versus placebo. Prescrit à la dose classique de 375 mg/m2/semaine pendant 4 semaines (étude scandinave) ou à une dose fixe de 1 g à J1 et J15, il permet d’obtenir un taux de réponse globale de 75 % à 1 an, versus 31 % pour le bras corticoïdes seul (avec ou sans placebo du rituximab). La tolérance globale est bonne, même si des accidents infectieux sévères et de rares cas de pneumocystose ont été rapportés, notamment chez des sujets âgés.6
En troisième ligne, en cas d’échec ou de rechute après rituximab, la splénectomie dont l’efficacité varie entre 50 et 75 % garde sa place mais, outre les risques infectieux bien connus, le risque accru de thrombose dans le contexte d’anémie hémolytique doit être pris en compte... Le rapport bénéfices-risques doit donc être bien pesé chez les patients ayant des antécédents de thrombose veineuse et/ou une thrombophilie avérée et notamment une biologie de type « antiphospholipide ». En cas de contre-indication à la chirurgie, l’embolisation splénique peut aussi donner de bons résultats, mais l’expérience se limite à des cas cliniques.8
Les immunosuppresseurs sont une alternative possible à la splénectomie, et bien sûr à privilégier en quatrième ligne en cas d’échec de la splénectomie. Aucune étude prospective et a fortiori contrôlée évaluant l’efficacité des différents immunosuppresseurs n’est disponible. Sur la base de quelques études rétrospectives et par analogie avec d’autres maladies auto-immunes, l’azathioprine à 2 mg/kg/j est le seul immunosuppresseur disposant d’une autorisation de mise sur le marché dans cette indication et est compatible avec la grossesse. En cas d’échec, les alternatives reposent sur le mycophénolate mofétil,9 la ciclosporine10 ou encore le sirolimus.11 Dans les formes les plus sévères, l’efficacité du cyclophosphamide à forte dose par voie intraveineuse, suivi ou non d’autogreffe, a également été rapportée.12
Le traitement de première ligne des formes « primaires » repose sur la corticothérapie à la dose initiale de 1 à 1,5 mg/kg/j à maintenir pendant 3 à 4 semaines suivie d’une décroissance progressive et, si possible, d’un arrêt en 3 à 6 mois. Ce traitement permet de guérir à lui seul environ un tiers des patients.6, 7
Le « dogme » ancien suggérant des doses plus fortes de corticoïdes et sur une durée prolongée (2 mg/kg initialement puis décroissance sur 2 ans) n’a plus de raison d’être car il ne reposait pas sur des études prospectives, et une durée prolongée de la corticothérapie ne diminue pas le taux de corticodépendance.
En deuxième ligne, en cas d’inefficacité primaire de la corticothérapie évaluée à 1 mois (rare) ou plus souvent de corticodépendance à haut niveau ou de rechute, le rituximab est privilégié. En effet, il a démontré son efficacité au travers de deux études prospectives randomisées dont une en double aveugle versus placebo. Prescrit à la dose classique de 375 mg/m2/semaine pendant 4 semaines (étude scandinave) ou à une dose fixe de 1 g à J1 et J15, il permet d’obtenir un taux de réponse globale de 75 % à 1 an, versus 31 % pour le bras corticoïdes seul (avec ou sans placebo du rituximab). La tolérance globale est bonne, même si des accidents infectieux sévères et de rares cas de pneumocystose ont été rapportés, notamment chez des sujets âgés.6
En troisième ligne, en cas d’échec ou de rechute après rituximab, la splénectomie dont l’efficacité varie entre 50 et 75 % garde sa place mais, outre les risques infectieux bien connus, le risque accru de thrombose dans le contexte d’anémie hémolytique doit être pris en compte... Le rapport bénéfices-risques doit donc être bien pesé chez les patients ayant des antécédents de thrombose veineuse et/ou une thrombophilie avérée et notamment une biologie de type « antiphospholipide ». En cas de contre-indication à la chirurgie, l’embolisation splénique peut aussi donner de bons résultats, mais l’expérience se limite à des cas cliniques.8
Les immunosuppresseurs sont une alternative possible à la splénectomie, et bien sûr à privilégier en quatrième ligne en cas d’échec de la splénectomie. Aucune étude prospective et a fortiori contrôlée évaluant l’efficacité des différents immunosuppresseurs n’est disponible. Sur la base de quelques études rétrospectives et par analogie avec d’autres maladies auto-immunes, l’azathioprine à 2 mg/kg/j est le seul immunosuppresseur disposant d’une autorisation de mise sur le marché dans cette indication et est compatible avec la grossesse. En cas d’échec, les alternatives reposent sur le mycophénolate mofétil,9 la ciclosporine10 ou encore le sirolimus.11 Dans les formes les plus sévères, l’efficacité du cyclophosphamide à forte dose par voie intraveineuse, suivi ou non d’autogreffe, a également été rapportée.12
Traitement des formes secondaires
Dans les formes secondaires d’anémie hémolytique auto-immune à anticorps chauds, en complément de la corticothérapie, qui reste la pierre angulaire du traitement de première ligne, certaines adaptations thérapeutiques peuvent être faites :
– en cas de lupus systémique : le rituximab a une excellente efficacité (taux de réponse 87,5 %),13 l’hydroxychloroquine peut avoir un intérêt dans le but d’une épargne cortisonique mais non démontré dans cette indication ;
– en cas de leucémie lymphoïde chronique sous-jacente : combinaison rituximab et chimiothérapie (schéma type : rituximab + cyclophosphamide + dexaméthasone, par exemple) en cas de leucémie évolutive ; éviter la fludarabine en monothérapie, les inhibiteurs de tyrosine kinase de Bruton (ibrutinib) dans les formes réfractaires, splénectomie à éviter ;
– en cas de déficit immunitaire commun variable (DICV) : les immunoglobulines polyvalentes à doses substitutives ne préviennent pas l’anémie hémolytique auto-immune. La splénectomie est à éviter. Le rituximab est efficace (taux de réponse 85 %) avec une tolérance acceptable dès lors que les patients sont substitués en immunoglobulines.14
En plus du traitement adapté à la maladie sous-jacente, une supplémentation en acide folique est systématique. Une anticoagulation préventive doit être proposée systématiquement en cas d’hospitalisation et peut même se discuter à domicile pendant les premières semaines de traitement et en phase d’hémolyse active.3 En cas de syndrome d’Evans avec une thrombopénie inférieure à 30 000/mm3, le bénéfice de l’anticoagulation n’est pas établi.
– en cas de lupus systémique : le rituximab a une excellente efficacité (taux de réponse 87,5 %),13 l’hydroxychloroquine peut avoir un intérêt dans le but d’une épargne cortisonique mais non démontré dans cette indication ;
– en cas de leucémie lymphoïde chronique sous-jacente : combinaison rituximab et chimiothérapie (schéma type : rituximab + cyclophosphamide + dexaméthasone, par exemple) en cas de leucémie évolutive ; éviter la fludarabine en monothérapie, les inhibiteurs de tyrosine kinase de Bruton (ibrutinib) dans les formes réfractaires, splénectomie à éviter ;
– en cas de déficit immunitaire commun variable (DICV) : les immunoglobulines polyvalentes à doses substitutives ne préviennent pas l’anémie hémolytique auto-immune. La splénectomie est à éviter. Le rituximab est efficace (taux de réponse 85 %) avec une tolérance acceptable dès lors que les patients sont substitués en immunoglobulines.14
En plus du traitement adapté à la maladie sous-jacente, une supplémentation en acide folique est systématique. Une anticoagulation préventive doit être proposée systématiquement en cas d’hospitalisation et peut même se discuter à domicile pendant les premières semaines de traitement et en phase d’hémolyse active.3 En cas de syndrome d’Evans avec une thrombopénie inférieure à 30 000/mm3, le bénéfice de l’anticoagulation n’est pas établi.
Anémies hémolytiques auto-immunes à anticorps froids
Elles sont souvent confondues à tort avec les cryoglobulinémies. Le tableau 3 résume les différences entre ces deux entités.
Les anémies hémolytiques auto-immunes à anticorps froids représentent 15 à 20 % des anémies hémolytiques auto-immunes. Le sex-ratio est de 1. Tous les âges peuvent être touchés (prédominance de formes post-infectieuses avant 50 ans et de maladies hématologiques après 50 ans).
L’auto-anticorps est systématiquement une IgM (kappa dans 90 % des cas) dirigée contre une cible à la surface du globule rouge (l’antigène I le plus souvent). La fixation de l’IgM est maximale au froid, donc au passage des hématies dans les capillaires des extrémités corporelles, et induit le recrutement de la fraction C3b du complément à la surface des globules rouges. Cela entraîne l’activation du complexe d’attaque membranaire du complément (hémolyse intravasculaire) et la reconnaissance par les macrophages spléniques des hématies opsonisées (hémolyse intratissulaire).
Les signes cliniques sont donc déclenchés par le froid ou par les situations augmentant le complément (fièvre, traumatisme, chirurgie). Ils associent des signes d’hémolyse intravasculaire (urines « porto », douleurs lombaires, fièvre) à un acrosyndrome (obstruction des microvaisseaux par des agglutinats d’hématies liées par des IgM).
Les tests biologiques, pratiqués à température ambiante, peuvent être perturbés par l’IgM. L’augmentation de la concentration corpusculaire moyenne en hémoglobine (CCMH) est très évocatrice. Elle est liée à un artéfact de mesure, l’automate considérant deux hématies agglutinées comme une seule. Ces tests se normalisent en chauffant les tubes à 37 °C.
Le TDA de routine est positif en complément (recruté par les IgM) et négatif en IgG.
Les maladies sous-jacentes sont de deux types :
– l’anémie hémolytique auto-immune à anticorps froids (« cold agglutinin syndrome ») post-infectieuse, qui touche l’enfant ou l’adulte jeune, fait suite à une primo-infection virale (virus d’Epstein-Barr, cytomégalovirus) ou à une infection bactérienne (mycoplasme). Elle guérit spontanément en quelques semaines avec l’infection et ne nécessite pas de traitement immunosuppresseur ;
– la maladie des agglutinines froides (« cold agglutinin disease »). Il s’agit d’une hémopathie B indolente (lymphome lymphoplasmocytique le plus souvent) où un clone médullaire produit l’IgM responsable de l’anémie hémolytique. Ce clone n’est habituellement pas responsable d’autres symptômes de lymphome (pas de syndrome tumoral, pas de signes généraux autres que ceux liés à l’anémie).
Les anémies hémolytiques auto-immunes à anticorps froids représentent 15 à 20 % des anémies hémolytiques auto-immunes. Le sex-ratio est de 1. Tous les âges peuvent être touchés (prédominance de formes post-infectieuses avant 50 ans et de maladies hématologiques après 50 ans).
L’auto-anticorps est systématiquement une IgM (kappa dans 90 % des cas) dirigée contre une cible à la surface du globule rouge (l’antigène I le plus souvent). La fixation de l’IgM est maximale au froid, donc au passage des hématies dans les capillaires des extrémités corporelles, et induit le recrutement de la fraction C3b du complément à la surface des globules rouges. Cela entraîne l’activation du complexe d’attaque membranaire du complément (hémolyse intravasculaire) et la reconnaissance par les macrophages spléniques des hématies opsonisées (hémolyse intratissulaire).
Les signes cliniques sont donc déclenchés par le froid ou par les situations augmentant le complément (fièvre, traumatisme, chirurgie). Ils associent des signes d’hémolyse intravasculaire (urines « porto », douleurs lombaires, fièvre) à un acrosyndrome (obstruction des microvaisseaux par des agglutinats d’hématies liées par des IgM).
Les tests biologiques, pratiqués à température ambiante, peuvent être perturbés par l’IgM. L’augmentation de la concentration corpusculaire moyenne en hémoglobine (CCMH) est très évocatrice. Elle est liée à un artéfact de mesure, l’automate considérant deux hématies agglutinées comme une seule. Ces tests se normalisent en chauffant les tubes à 37 °C.
Le TDA de routine est positif en complément (recruté par les IgM) et négatif en IgG.
Les maladies sous-jacentes sont de deux types :
– l’anémie hémolytique auto-immune à anticorps froids (« cold agglutinin syndrome ») post-infectieuse, qui touche l’enfant ou l’adulte jeune, fait suite à une primo-infection virale (virus d’Epstein-Barr, cytomégalovirus) ou à une infection bactérienne (mycoplasme). Elle guérit spontanément en quelques semaines avec l’infection et ne nécessite pas de traitement immunosuppresseur ;
– la maladie des agglutinines froides (« cold agglutinin disease »). Il s’agit d’une hémopathie B indolente (lymphome lymphoplasmocytique le plus souvent) où un clone médullaire produit l’IgM responsable de l’anémie hémolytique. Ce clone n’est habituellement pas responsable d’autres symptômes de lymphome (pas de syndrome tumoral, pas de signes généraux autres que ceux liés à l’anémie).
Traitement
Le traitement doit systématiquement contenir une supplémentation en folates et une éviction du froid (port de gants, bonnet, écharpe). En cas de transfusion, on utilise un réchauffeur de culot globulaire. Pour éviter les transfusions, on peut utiliser l’érythropoïétine.
La cortisone est insuffisamment efficace pour être recommandée. La splénectomie est à éviter car elle ne traite pas la part intravasculaire de l’hémolyse.
Pour les patients très symptomatiques et nécessitant des transfusions, on cible donc le clone producteur de l’IgM. Une équipe norvégienne est à l’origine des trois études multicentriques, prospectives, non contrôlées sur la thérapeutique des maladies des agglutinines froides : rituximab seul,15 rituximab et fludarabine,16 rituximab et bendamustine.17 Letableau 4 en résume les résultats. L’association rituximab-bendamustine semble la plus efficace sur le long terme, au prix d’effets indésirables hématologiques non négligeables.
Compte tenu de la physiopathologie (l’IgM active fortement le complément), les inhibiteurs du complément constituent une classe prometteuse. L’éculizumab, anticorps anti-C5a, a été utilisé dans une étude allemande prospective en ouvert, non contrôlée portant sur 13 patients et a permis une nette diminution des besoins transfusionnels mais pas d’effets sur les symptômes circulatoires.18 Le sutimlimab, anticorps anti-C1, est actuellement à l’étude.19
La cortisone est insuffisamment efficace pour être recommandée. La splénectomie est à éviter car elle ne traite pas la part intravasculaire de l’hémolyse.
Pour les patients très symptomatiques et nécessitant des transfusions, on cible donc le clone producteur de l’IgM. Une équipe norvégienne est à l’origine des trois études multicentriques, prospectives, non contrôlées sur la thérapeutique des maladies des agglutinines froides : rituximab seul,15 rituximab et fludarabine,16 rituximab et bendamustine.17 Le
Compte tenu de la physiopathologie (l’IgM active fortement le complément), les inhibiteurs du complément constituent une classe prometteuse. L’éculizumab, anticorps anti-C5a, a été utilisé dans une étude allemande prospective en ouvert, non contrôlée portant sur 13 patients et a permis une nette diminution des besoins transfusionnels mais pas d’effets sur les symptômes circulatoires.18 Le sutimlimab, anticorps anti-C1, est actuellement à l’étude.19
Conclusion
Les anémies hémolytiques auto-immunes sont des maladies rares dont la mortalité n’est pas négligeable. La détermination de l’optimum thermique de l’auto-anticorps (anticorps chauds ou froids) est capitale puisqu’elle détermine le bilan causal à effectuer et le traitement (tableau 5 ). Pour les anémies hémolytiques auto-immunes à anticorps chauds, il n’y a plus de place pour une corticothérapie prolongée, le rituximab intervenant rapidement dans la stratégie thérapeutique.
* Le volume globulaire moyen se mesure en femtolitres (fL = 10-15 L).
Remerciements au Pr Marc Michel, pour les discussions sur les anémies hémolytiques auto-immunes et les conseils pour ce travail.
Références
1. Xu Y, Yang W, Liao L, et al. Mean reticulocyte volume: a specific parameter to screen for hereditary spherocytosis. Eur J Haematol 2016;96:170‑4.
2. Hendrick AM. Auto-immune haemolytic anaemia--a high-risk disorder for thromboembolism. Hematology 2003;8:53‑6.
3. Audia S, Bach B, Samson M, et al. Venous thromboembolic events during warm autoimmune hemolytic anemia. Garcia de Frutos P (ed). PLoS ONE 2018;13:e0207218.
4. Roumier M, Loustau V, Guillaud C, et al. Characteristics and outcome of warm autoimmune hemolytic anemia in adults: new insights based on a single-center experience with 60 patients. Am J Hematol 2014;89:E150-5.
5. Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood 2014;124:2930‑6.
6. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study): rituximab for wAIHA in adults. Am J Hematol 2017;92:23‑7.
7. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol 2013;163:393‑9.
8. Molica M, Massaro F, Annechini G, et al. Life-threatening autoimmune hemolytic anemia and idhiopatic thrombocytopenic purpura. Successful selective splenic artery embolization. Mediterr J Hematol Infect Dis. 2016;8:e2016020.
9. Howard J, Hoffbrand AV, Prentice HG, Mehta A. Mycophenolate mofetil for the treatment of refractory auto-immune haemolytic anaemia and auto-immune thrombocytopenia purpura. Br J Haematol 2002;117:712‑5.
10. Janić D, Krivokapić-Dokmanović L, Jovanović N, Lazić J, Rodić P, Janković S. Glucocorticoid-resistant Evans’ syndrome successfully controlled with low-dose cyclosporine. Int J Clin Pharmacol Ther 2011;49:622‑5.
11. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420‑4.
12. Panceri R, Fraschini D, Tornotti G, Masera G, Locasciulli A, Bacigalupo A. Successful use of high-dose cyclophosphamide in a child with severe autoimmune hemolytic anemia. Haematologica 1992;77:76‑8.
13. Serris A, Amoura Z, Canouï-Poitrine F, et al. Efficacy and safety of rituximab for systemic lupus erythematosus-associated immune cytopenias: A multicenter retrospective cohort study of 71 adults. Am J Hematol 2018;93:424‑9.
14. Gobert D, Bussel JB, Cunningham-Rundles C, et al. Efficacy and safety of rituximab in common variable immunodeficiency-associated immune cytopenias: a retrospective multicentre study on 33 patients. Br J Haematol 2011;155:498‑508.
15. Berentsen S, Ulvestad E, Gjertsen BT, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood 2004;103:2925‑8.
16. Berentsen S, Randen U, Vågan AM, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood 2010;116:3180‑4.
17. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood 2017;130:537‑41.
18. Röth A, Bommer M, Hüttmann A, et al. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv 2018;2:2543‑9.
19. Berentsen S, Röth A, Randen U, Jilma B, Tjønnfjord GE. Cold agglutinin disease: current challenges and future prospects. J Blood Med 2019;10:93‑103.
2. Hendrick AM. Auto-immune haemolytic anaemia--a high-risk disorder for thromboembolism. Hematology 2003;8:53‑6.
3. Audia S, Bach B, Samson M, et al. Venous thromboembolic events during warm autoimmune hemolytic anemia. Garcia de Frutos P (ed). PLoS ONE 2018;13:e0207218.
4. Roumier M, Loustau V, Guillaud C, et al. Characteristics and outcome of warm autoimmune hemolytic anemia in adults: new insights based on a single-center experience with 60 patients. Am J Hematol 2014;89:E150-5.
5. Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood 2014;124:2930‑6.
6. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study): rituximab for wAIHA in adults. Am J Hematol 2017;92:23‑7.
7. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol 2013;163:393‑9.
8. Molica M, Massaro F, Annechini G, et al. Life-threatening autoimmune hemolytic anemia and idhiopatic thrombocytopenic purpura. Successful selective splenic artery embolization. Mediterr J Hematol Infect Dis. 2016;8:e2016020.
9. Howard J, Hoffbrand AV, Prentice HG, Mehta A. Mycophenolate mofetil for the treatment of refractory auto-immune haemolytic anaemia and auto-immune thrombocytopenia purpura. Br J Haematol 2002;117:712‑5.
10. Janić D, Krivokapić-Dokmanović L, Jovanović N, Lazić J, Rodić P, Janković S. Glucocorticoid-resistant Evans’ syndrome successfully controlled with low-dose cyclosporine. Int J Clin Pharmacol Ther 2011;49:622‑5.
11. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420‑4.
12. Panceri R, Fraschini D, Tornotti G, Masera G, Locasciulli A, Bacigalupo A. Successful use of high-dose cyclophosphamide in a child with severe autoimmune hemolytic anemia. Haematologica 1992;77:76‑8.
13. Serris A, Amoura Z, Canouï-Poitrine F, et al. Efficacy and safety of rituximab for systemic lupus erythematosus-associated immune cytopenias: A multicenter retrospective cohort study of 71 adults. Am J Hematol 2018;93:424‑9.
14. Gobert D, Bussel JB, Cunningham-Rundles C, et al. Efficacy and safety of rituximab in common variable immunodeficiency-associated immune cytopenias: a retrospective multicentre study on 33 patients. Br J Haematol 2011;155:498‑508.
15. Berentsen S, Ulvestad E, Gjertsen BT, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood 2004;103:2925‑8.
16. Berentsen S, Randen U, Vågan AM, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood 2010;116:3180‑4.
17. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood 2017;130:537‑41.
18. Röth A, Bommer M, Hüttmann A, et al. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv 2018;2:2543‑9.
19. Berentsen S, Röth A, Randen U, Jilma B, Tjønnfjord GE. Cold agglutinin disease: current challenges and future prospects. J Blood Med 2019;10:93‑103.
Dans cet article
- Quelle démarche diagnostique ?
- Tests biologiques utiles pour le diagnostic
- Certains tests ne sont pas nécessaires en pratique clinique de routine
- Épidémiologie et classification des anémies hémolytiques auto-immunes
- Anémies hémolytiques auto-immunes à anticorps chauds
- Anémies hémolytiques auto-immunes à anticorps froids
- Conclusion



Résumé
Les anémies hémolytiques auto-immunes sont une cause rare d’anémie hémolytique acquise, liée à la présence d’un auto-anticorps dirigé contre un ou plusieurs antigènes exprimé(s) à la surface du globule rouge et le plus souvent attestée par un test direct à l’antiglobuline (TDA) positif. Rarement, les anémies hémolytiques auto-immunes peuvent être arégénératives (10-20 % des cas), et le TDA peut être négatif (5 % des cas). On en distingue deux formes principales : les anémies hémolytiques auto-immunes à anticorps chauds (TDA positif de type IgG +/- C3d) et les anémies hémolytiques auto-immunes à anticorps froids (TDA positif de type C3d), qui diffèrent par leurs causes sous-jacentes et par leur traitement. Les anémies hémolytiques auto-immunes à anticorps chauds sont dans 50 % des cas « secondaires » à une hémopathie lymphoïde B, un déficit immunitaire commun variable, un lupus systémique, ou induites par un médicament ; le traitement repose sur une corticothérapie courte (3 à 6 mois), et le rituximab est le traitement de 2e ligne. Les anémies hémolytiques auto-immunes à anticorps froids sont de deux types : soit postinfectieuses (mycoplasme, virus d’Epstein-Barr), soit liées à une maladie des agglutinines froides, qui est une hémopathie clonale B indolente ; le traitement est avant tout symptomatique et repose sur les mesures de protection vis-à-vis du froid. La corticothérapie et la splénectomie sont inefficaces. En cas d’anémie marquée, un traitement par rituximab seul ou combiné à une chimiothérapie est indiqué.