Quels signes d’appel ?
Une pathologie complexe et évolutive
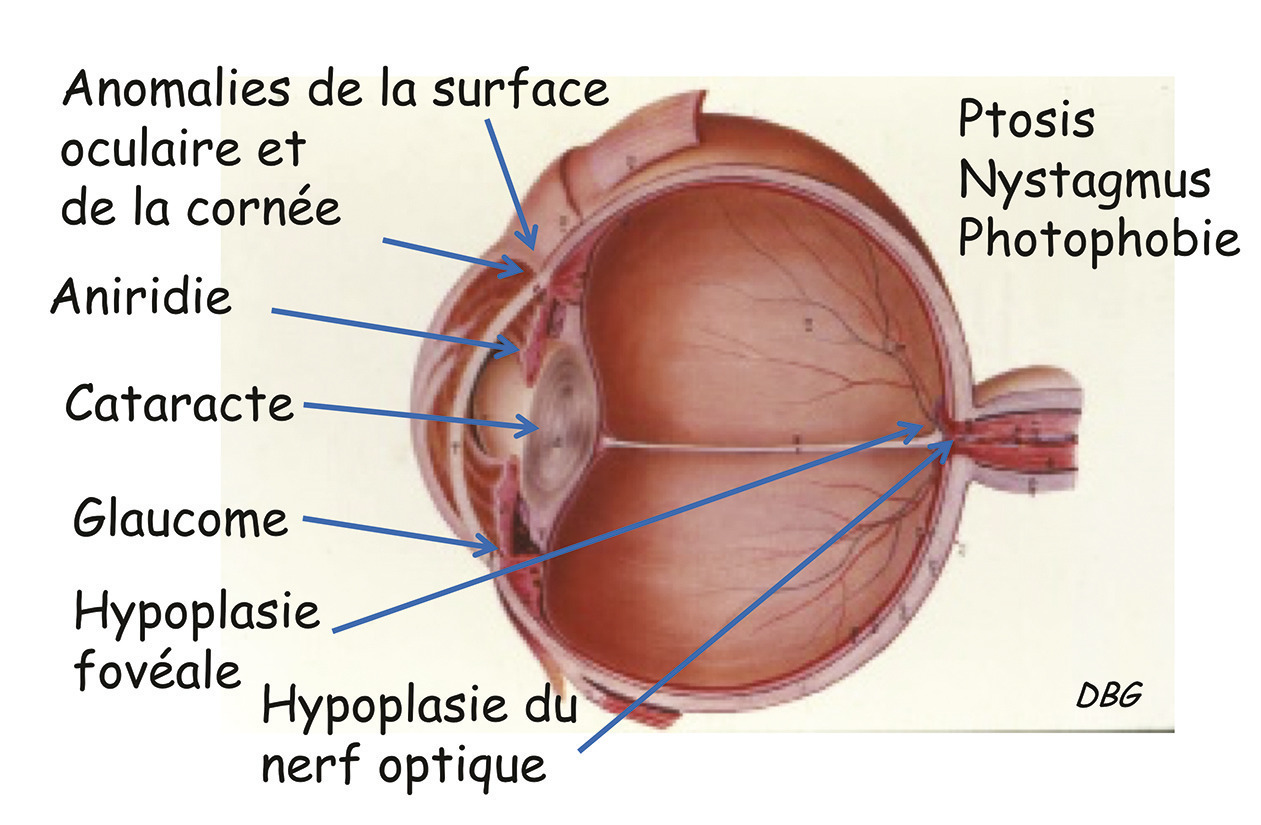
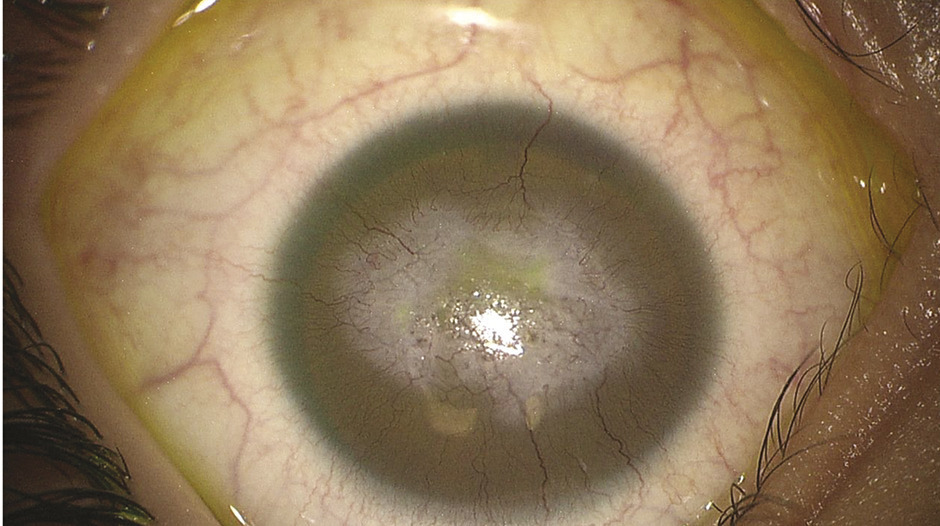
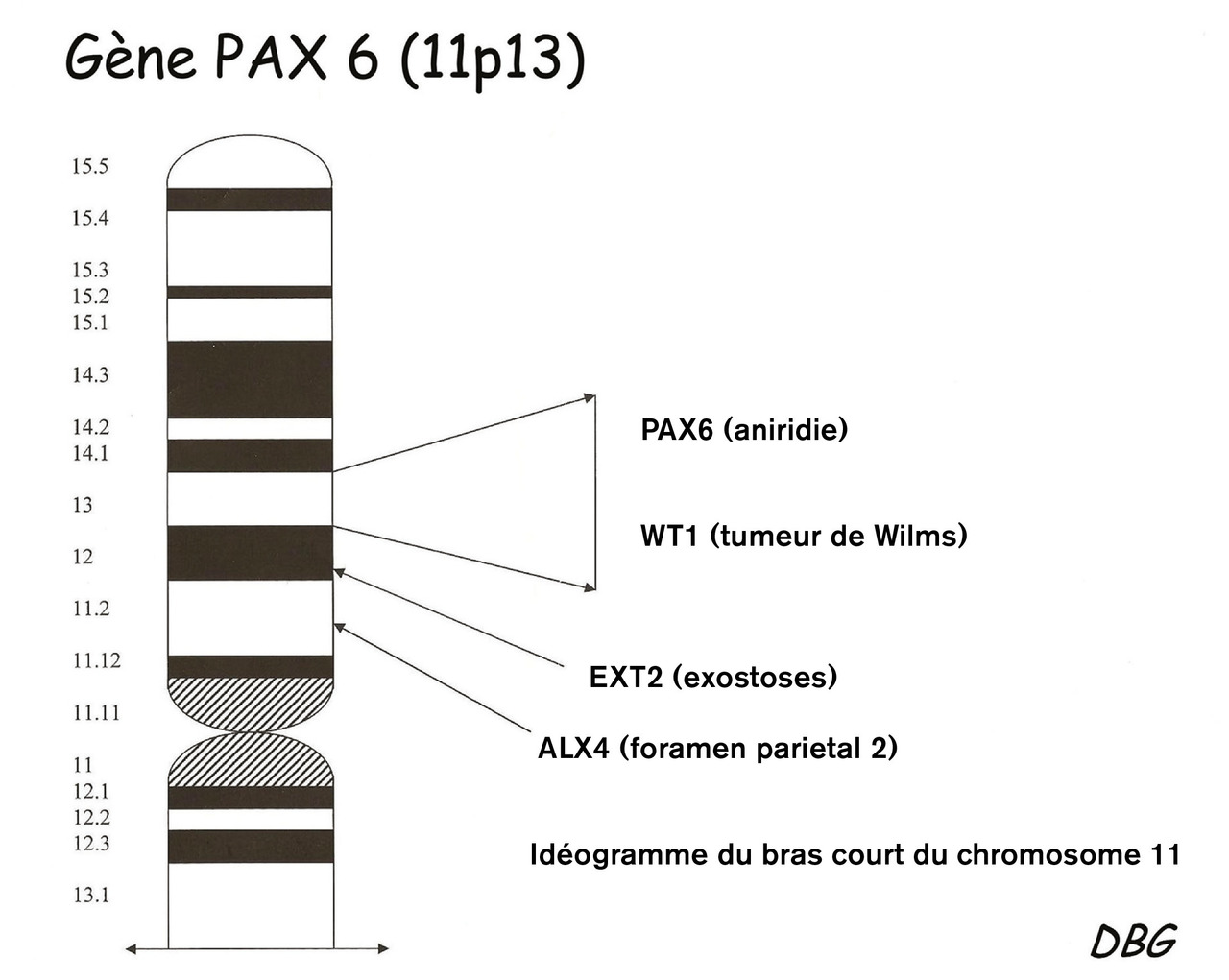
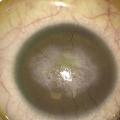
L’atteinte cornéenne à type de kératopathie fréquente est due à une insuffisance limbique évolutive depuis l’enfance responsable d’opacités variables. Elle nécessite la prescription d’agents mouillants sans conservateurs, cependant les opacités cornéennes peuvent s’aggraver jusqu’à une cornée blanche et néovascularisée malgré les nombreuses options thérapeutiques employées (fig. 3).
Le glaucome est l’une des complications les plus fréquentes de l’aniridie, due à l’anomalie de l’angle irido- cornéen qui réduit la résorption de l’humeur aqueuse en entraînant une élévation de la pression intra-oculaire (PIO). La mesure de la pression doit être réalisée régulièrement pour dépister précocement le glaucome et le traiter.
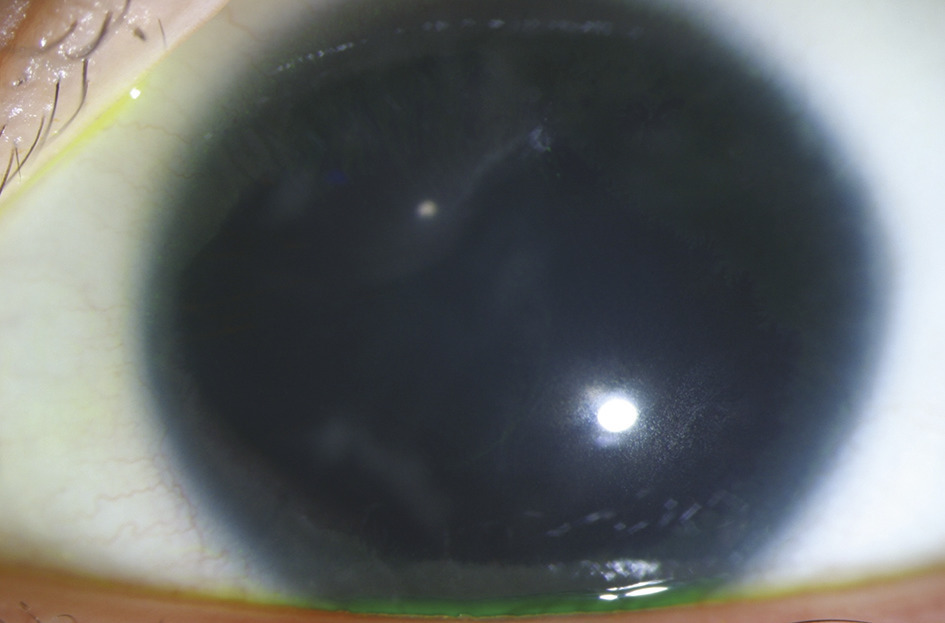
L’opacification du cristallin est souvent limitée à des opacités non obturantes chez l’enfant revêtant plus rarement une forme de cataracte congénitale obturante. Lors de l’apparition d’une cataracte obturante, une chirurgie du cristallin à type de phaco-exérèse est nécessaire mais en connaissant les risques accrus de complications lors de la chirurgie et de la décompensation de la cornée qui peut en découler. L’implant cristallinien placé est classique sans diaphragme irien qui augmente les risques de glaucome.
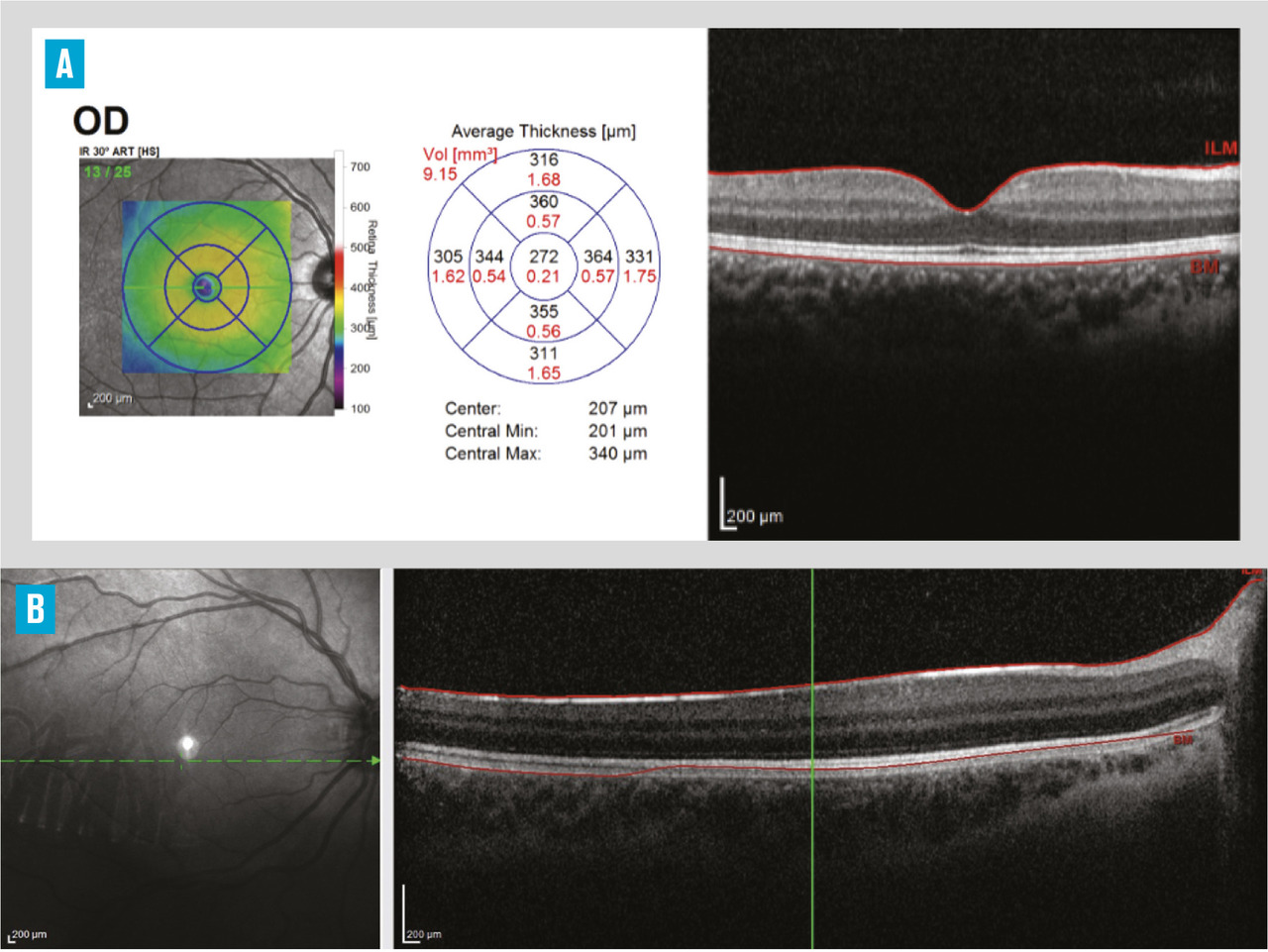
L’hypoplasie fovéolaire, quasi systématique, est évaluée grâce à une tomographie en cohérence optique (OCT) [fig. 4].
Lorsque le nystagmus et la transparence cornéenne le permettent, de nombreux examens complémentaires ophtalmologiques peuvent être réalisés pour mieux caractériser la maladie tels, de façon non exhaustive, que : champ visuel, OCT antérieure et postérieure, vidéotopographie, pachymétrie, ultrasound biomicroscopy (UBM), rétinographie, rétinographie grand champ. Ils permettront aussi de suivre la maladie et ses complications de façon fiable.
Chez l’enfant, un examen à pratiquer sous anesthésie générale est nécessaire pour effectuer un phénotypage complet et dépister les complications potentielles.
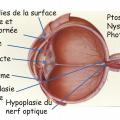
La déficience visuelle et la photophobie entraînent un handicap visuel avec des difficultés pour la mobilité, les déplacements, la communication, les apprentissages, la motricité fine, l’autonomie, avec des conséquences dans la vie scolaire, professionnelle, socioculturelle et sportive. La rééducation fonctionnelle et certaines aides techniques peuvent pallier ces situations de handicap en apportant une meilleure qualité de vie au patient. L’évolution de la recherche clinique sur les cellules souches cornéennes et la thérapie génique permettent d’espérer de mieux traiter l’atteinte oculaire et d’améliorer la qualité de vie des patients.3
Y a-t-il d’autres anomalies ?
Chez certains patients des anomalies systémiques peuvent être associées. Des anomalies cérébrales incluant le corps calleux et le système auditif et olfactif sont parfois observées ; elles doivent alors être évaluées si possible par un neuro- pédiatre. Chez l’adulte, le risque de diabète précoce est plus élevé et impose une surveillance régulière de la glycémie.
Que doit-on éliminer ?
Syndrome WAGR
Syndrome de Gillespie
Quel bilan génétique ?
OPTIMISER LE DIAGNOSTIC
Centres de référence des maladies rares en ophtalmologie
L’aniridie congénitale est une maladie rare qui nécessite un bilan dans un centre de référence des maladies rares en ophtalmologie. La filière SENSGENE regroupe en France cinq centres de référence des maladies rares en ophtalmologie qui coordonnent des centres constitutifs et des centres de compétences répartis sur le territoire français métropolitain et outremer. Certains centres de référence européens sont aussi reliés par le réseau européen European reference network on rare eye diseases (ERN). Le centre de référence détermine la fréquence, les modalités et le médecin référent pour optimiser la prise en charge de l’aniridie, la prévention des complications secondaires et la surveillance. Une transition de la prise en charge enfant-adolescent-adulte est souvent nécessaire. OPHTARA Centre de Maladies Rares en Ophtalmologie et Centre Européen ERN EYE, est un des 5 centres de la filière.
2. Lee HJ, Colby KA. A review of the clinical and genetic aspects of aniridia. Semin Ophthalmol 2013;28:306-12.
3. Bremond-Gignac D, Copin H, Benkhalifa M. Corneal epithelial stem cells for corneal injury. Expert Opin Biol Ther 2018;9:1-7.
4. Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet 2012;20:1011-7.
5. Gerber S, Alzayady KJ, Burglen L, et al. Recessive and dominant de novo ITPR1 mutations cause Gillespie syndrome. Am J Hum Genet 2016;98:971-80.
OPHTARA, centre de référence des maladies rares en ophtalmologie, filière SENSGENE, Centre rare eye disease european rare disease network.
PNDS sur l’aniridie en cours de rédaction qui sera disponible sur le site de la Haute Autorité de santé.
http://www.maladiesrares-necker.aphp.fr/ophtara/
http://www.sensgene.com/les-centres-de-reference/ophtara-centre-de-reference-maladies-rares-en-ophtalmologie
GENIRIS, association nationale loi 1901, sur l’aniridie et les pathologies rares de l’iris avec ou sans syndromes associés www.geniris.fr/
ANIRIDIA EUROPE, European non-governmental and non-profit federation on aniridia.
https://www.aniridia.eu
 Encadrés
Encadrés