Deux grandes catégories : les tumeurs et les malformations. Depuis quelques années, les mutations génétiques causales sont progressivement identifiées, permettant une meilleure compréhension de la physiopathologie et des progrès thérapeutiques.
Il s’agit d’un groupe hétérogène de pathologies développées aux dépens des vaisseaux de tous types. La classification de l’International Society for the Study of Vascular Anomalies (ISSVA) adoptée depuis 1992 et régulièrement actualisée, est fondée sur les caractéristiques cliniques, radiologiques, histologiques, biologiques et maintenant moléculaires.1
Elle distingue 2 groupes : d’une part, les tumeurs qui résultent d’une hyperplasie cellulaire, avec au premier rang l’hémangiome infantile ; d’autre part, les malformations, présentes dès la naissance mais pas toujours apparentes à ce moment-là. Ces dernières peuvent toucher tous les types de vaisseaux et parfois s’associer pour réaliser des tableaux de malformations vasculaires complexes.
Hémangiome infantile (HI)
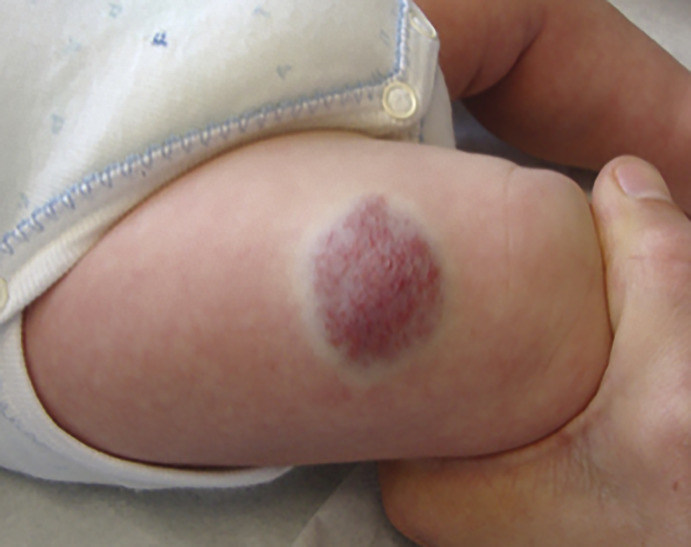
C’est la tumeur vasculaire la plus fréquente chez l’enfant, touchant 5 à 10 % des bébés après la naissance. Elle peut apparaître n’importe où sur le tégument, y compris sur les muqueuses. Les facteurs de risque sont la grande prématurité, un poids de naissance inférieur à 1 500 g, les anomalies placentaires (éclampsie, procédures invasives), le sexe féminin, un âge maternel élevé, une grossesse multiple. Les formes sévères atteignent plus volontiers les filles.
Qu’il soit superficiel (
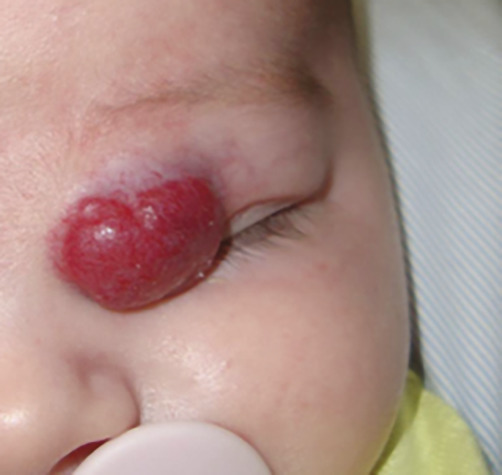
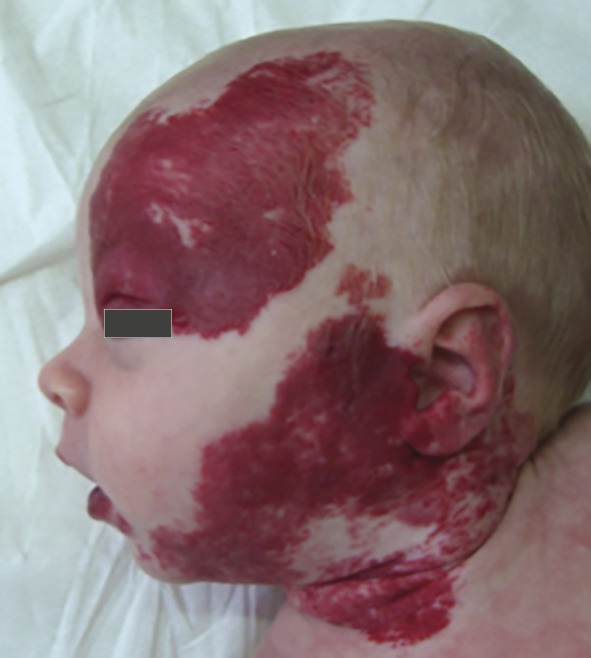
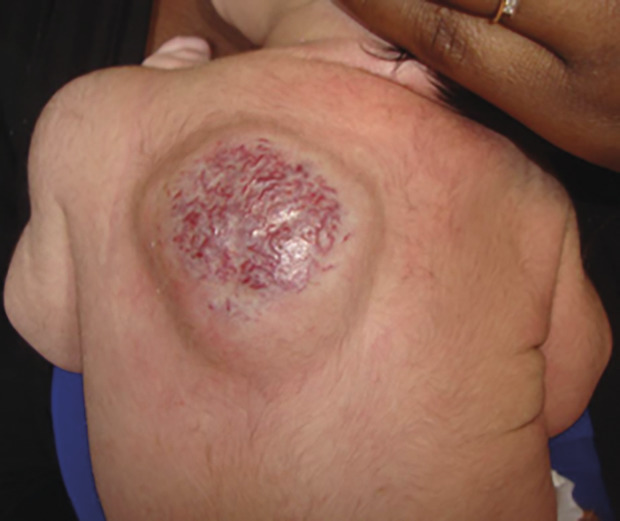
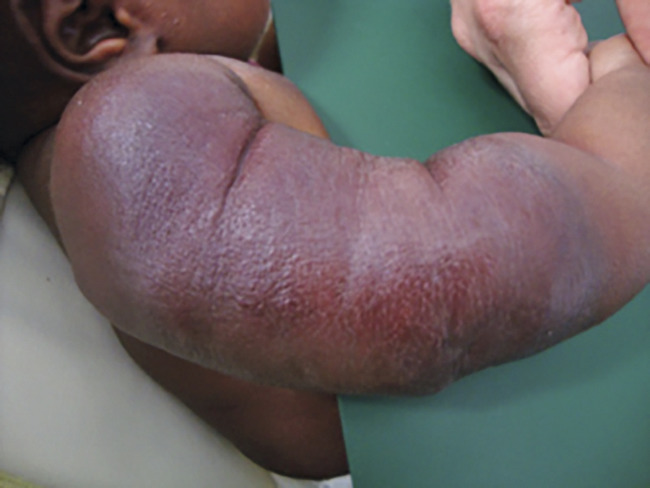
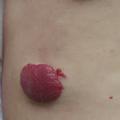
Superficiel, il a un aspect caractéristique : masse papuleuse plus ou moins ferme, bien limitée, rouge vif ou framboise, avec une surface volontiers mamelonnée. Localisé en sous-cutané, c’est une voussure bleutée, moins spécifique (
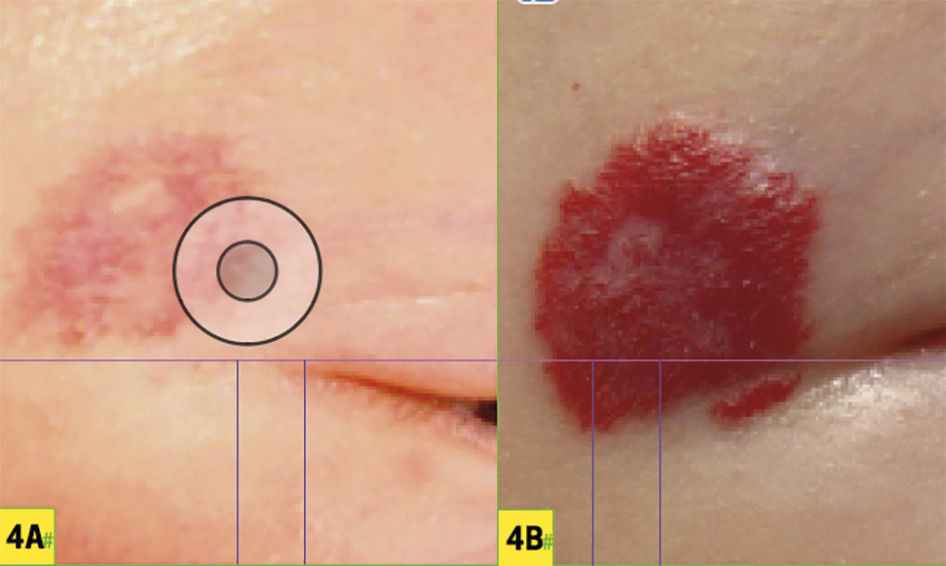
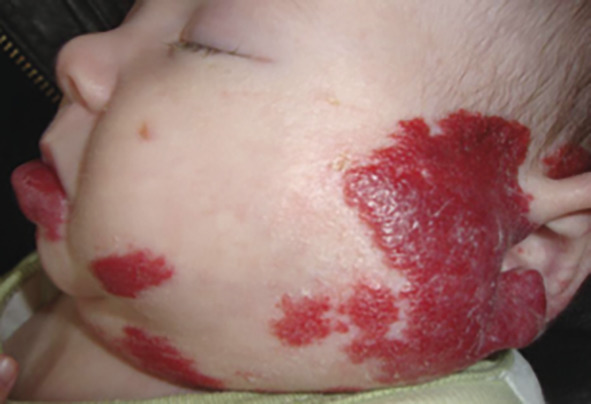
Les formes segmentaires, c’est-à-dire en nappe, sont parfois difficiles à différencier des angiomes plans : une lésion vasculaire plane qui fonce avec le temps, rapidement évolutive, est une anomalie proliférative et donc un HI.
Ces hémangiomes, certes bénins, sont accompagnés dans 15-20 % d’une morbidité non négligeable : gêne esthétique, fonctionnelle, ulcération, voire risque vital, ou encore association à des anomalies malformatives (lésions segmentaires syndromiques). Tout nourrisson ayant une forme à risque (
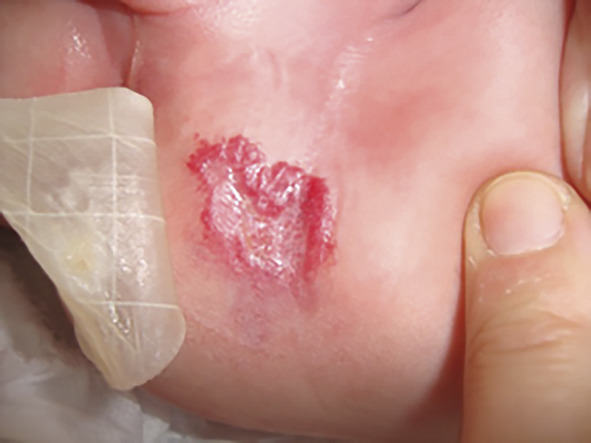
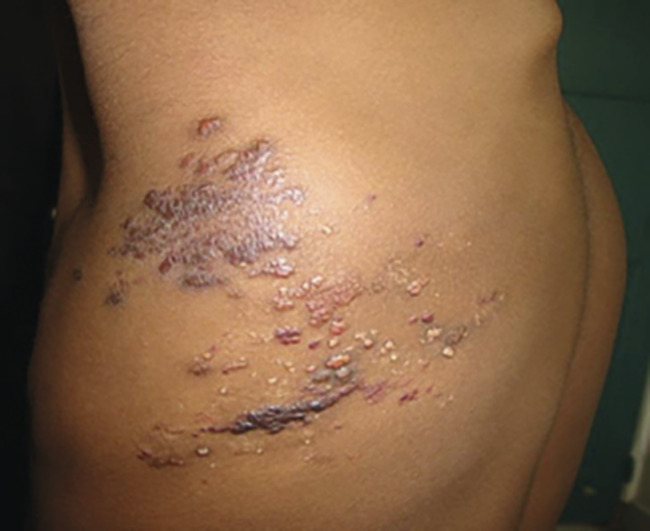
L’ulcération survient surtout vers les 2e et 3e mois d’évolution; c’est-à-dire en phase proliférative, mais parfois plus tardivement. Douloureuse, elle n’entraîne qu’exceptionnellement des saignements importants responsables d’une anémie. Elle complique le plus souvent les HI de grande taille, notamment segmentaires, et ceux situés sur des zones de macération et d’humidité (siège +++ ;
Le retentissement fonctionnel est lié au caractère obstructif des HI péri-orificiels volumineux : œil, bouche, nez, oreille. Ceux siégeant dans la région orbitaire imposent une évaluation ophtalmologique systématique. Une occlusion palpébrale complète peut être responsable d’une amblyopie si elle n’est pas levée (
Le risque vital est rare : une gêne voire une détresse respiratoire sont possibles en cas de localisation au niveau des voies aériennes supérieures, fréquemment associée aux HI cutanés du territoire mandibulaire (dit en « barbe »), parfois restreints à la lèvre inférieure (
Les HI segmentaires, lorsqu’ils sont situés dans la région céphalique (syndrome PHACES ;
Les HI ne se compliquent jamais d’anomalie de la coagulation. ,
Actuellement, pour les formes compliquées, le propranolol (Hemangiol), à la posologie de 3 mg/kg/j, à commencer entre 5 semaines et 5 mois d’âge,2 a une efficacité spectaculaire : après 1 mois de traitement, 90 % des enfants sont améliorés ; dans 60 % des cas, l’HI régresse complètement après 6 mois ou il ne persiste que quelques télangiectasies. La mise en route (à doses progressives) est faite en milieu pédiatrique (surveillance de la fréquence cardiaque et de la PA après la 1re prise) puis le traitement est ambulatoire.
Hémangiomes congénitaux
Tumeurs strictement congénitales, elles sont visibles dès la naissance, à leur taille maximale, sans phase de croissance post-natale. Elles sont beaucoup moins fréquentes que les HI, sans facteur favorisant ; le sex-ratio est équilibré. Caractéristiques : halo anémique très marqué, télangiectasies d’aspect variable.
Trois sous-types sont décrits, selon l’évolution clinique :
– le RICH (Rapidly Involuting Congenital Hemangioma), grosse tumeur saillante qui involue spontanément de façon rapide, en général en 6 à 12 mois (
– le NICH (Non Involuting Congenital Hemangioma), d’emblée plan qui persiste tout au long de la vie (
– et le PICH (Partially Involuting Congenital Hemangioma), qui commence comme un RICH n’involuant que partiellement et laissant une lésion résiduelle de type NICH. Les complications sont rares : ulcération pouvant provoquer un syndrome hémorragique ; thrombopénie transitoire et hyperdébit, voire insuffisance cardiaque pour les atteintes volumineuses.
Le diagnostic est clinique. L’imagerie, en particulier l’IRM (caractéristiques très voisines de celles de l’HI) est réalisée à visée préthérapeutique, si on envisage une embolisation ou une chirurgie, dans les cas compliqués ou dans un but esthétique. Dans certaines formes atypiques, le diagnostic différentiel de fibrosarcome congénital doit être écarté par une biopsie profonde.
Syndrome de Kasabach-Merritt
Le phénomène de Kasabach-Merritt (PKM), extrêmement rare, est caractérisé par la survenue brutale d’une thrombopénie profonde accompagnée d’une coagulation intravasculaire disséminée (CIVD) d’intensité variable, chez un nourrisson atteint d’une tumeur vasculaire bien particulière : l’hémangio- endothéliome kaposiforme et l’angiome en touffes (
La tumeur est infiltrante, tendue, luisante, érythémateuse ou violacée, souvent ecchymotique, douloureuse, parfois compressive – selon la localisation –, pouvant induire une insuffisance cardiaque à haut débit. Les anomalies de l’hémostase peuvent entraîner des manifestations hémorragiques.
Le PKM survient dès l’installation de la tumeur ou après un intervalle libre de durée variable, de quelques semaines ou mois, sans facteur déclenchant identifiable.
La phase aiguë est caractérisée par une thrombopénie profonde, un fibrinogène très bas, parfois indosable, une élévation des D-dimères. Le TCA est allongé et le TP diminué, mais de façon inconstante.
à distance de l’épisode, les lésions cutanées rédiduelles, quasi constantes, peuvent être le siège de poussées inflammatoires, avec parfois des modifications mineures de l’hémostase, et s’accompagner de séquelles fonctionnelles.
Le sirolimus est efficace dans la phase aiguë, mais il doit être maintenu de façon prolongée.3
Malformations vasculaires
On distingue celles à flux lent (capillaire, veineuse, lymphatique) de celles à flux rapide (artérioveineuse). Dans la plupart des cas, la mutation génétique causale est identifiée (
Angiome plan (AP)
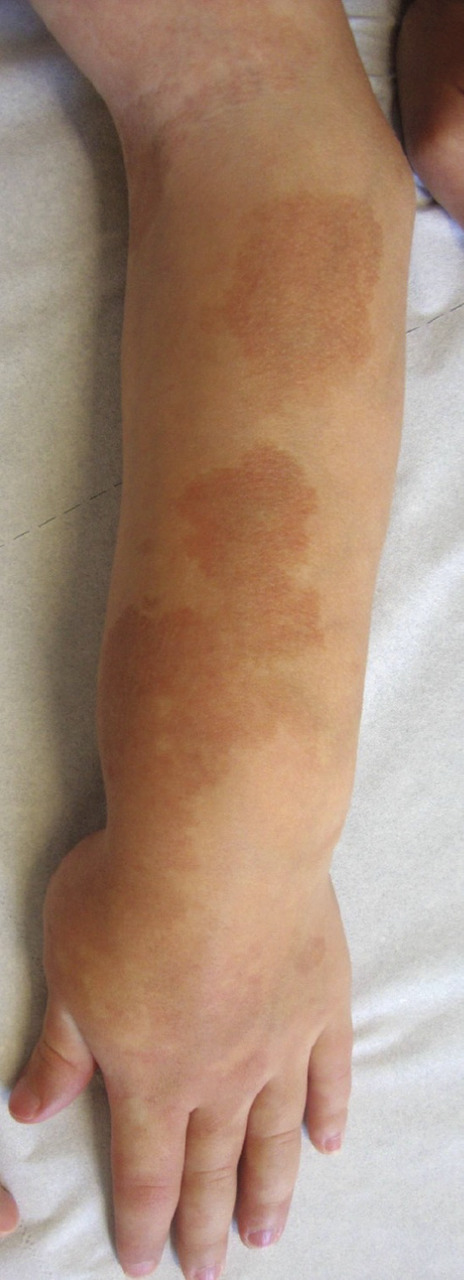
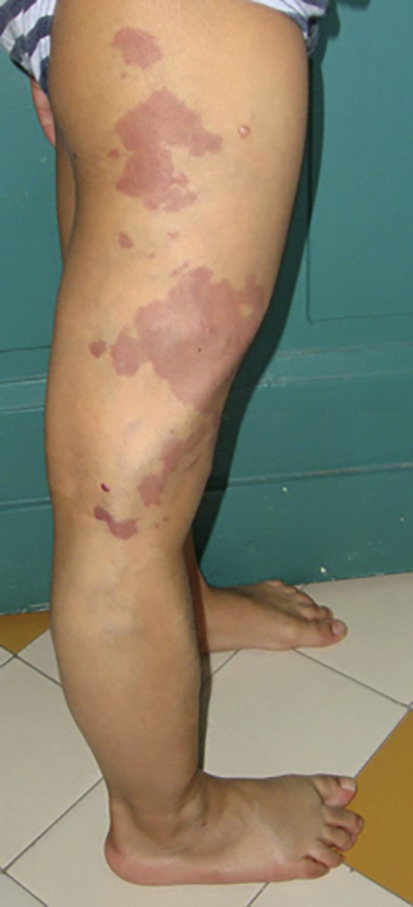
Malformation congénitale faite de vaisseaux capillaires ectasiques dans le derme, son diagnostic est clinique : macule érythémateuse qui pâlit durant le premier mois, et croît avec le temps (
En revanche, devant une atteinte du visage (territoire frontal), il faut rechercher un syndrome de Sturge-Weber, qui associe des anomalies ophtalmiques par angiomatose choroïdienne pouvant entraîner un glaucome (urgence thérapeutique), des manifestations neurologiques liées à une angiomatose méningée (épilepsie précoce, retard mental, syndrome déficitaire ;
En cas de gêne esthétique, on peut proposer le laser vasculaire colorant pulsé qui éclaircit ces taches de vin (mais de nombreuses séances sont nécessaires).
Malformations veineuses
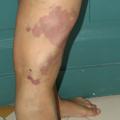
Elles peuvent siéger n’importe où sur le corps, dans les tissus sous-cutanés, les muscles et les muqueuses. Ce sont des masses ou nappes bleutées gonflant en déclivité ou à l’effort, se vidant par compression (
Principale symptomatologie : la douleur, en rapport avec les événements thrombo-inflammatoires survenant à cause de la stase sanguine. Celle-ci est responsable d’une activation chronique de la coagulation ou coagulation intravasculaire localisée (CIVL) dont la sévérité est corrélée au degré d’extension, en particulier musculaire, autrement dit à la masse lésionnelle.
Le marqueur biologique de cette activation est l’élévation des D-dimères.5 Des taux > 1 800 ng/mL et un fibrogène bas imposent un traitement par HBPM avant tout geste chirurgical.
Les formes multifocales sont rares (
Malformations lymphatiques kystiques
Elles sont macrokystiques, microkystiques ou mixtes ; superficielles ou profondes, cutanées, muqueuses, rarement viscérales, mono- ou multikystiques.
Dans les formes macrokystiques volumineuses, le diagnostic est soit anténatal, soit fait à la naissance (le plus souvent), et dans 80 à 90 % des cas avant l’âge de 2 ans.
Macrokystique, c’est une tuméfaction ronde, globuleuse, sous une peau de couleur normale ou parfois rose lilacé, généralement lisse, de consistance ferme et élastique, ou molle et dépressible. Elle est parfois associée à une composante superficielle : des vésicules correspondant à des lymphangiectasies de surface.
Les formes purement microkystiques sont des plaques constituées de vésicules à contenu clair ou hématique (
Principales complications : risque de compression des organes de voisinage (voies aériennes supérieures +++), et poussées inflammatoires douloureuses, accompagnant souvent une infection virale, et/ou hémorragie intrakystique, le plus souvent spontanée. La lésion devient alors rouge, tendue, puis ecchymotique, et son volume peut nettement augmenter.
L’imagerie, en particulier l’IRM, évaluant l’extension locale, avant discussion thérapeutique, montre des kystes de taille variable, hypoT1, hyper T2, ne prenant pas le contraste.
Malformations artérioveineuses
Elles sont les plus rares des malformations vasculaires superficielles. La plupart sont situées dans la région cervico-faciale. Elles sont formées de vaisseaux artériels et veineux dysmorphiques interconnectés directement (shunts), sans capillaires intermédiaires, formant une structure vasculaire appelée nidus. Dans l’enfance, elles ont l’aspect d’une tache rouge et chaude. La puberté et les traumatismes favorisent leur évolution, avec apparition de douleur, thrill, hyperhidrose, hypertrichose et hyperthermie.
L’association à des malformations capillaires multiples de petite taille non systématisées, caractérisées par un halo clair et une teinte allant du rosé au brun clair, évoque un syndrome CM-AVM (capillary malformation-arteriovenous malformation), pathologie héréditaire (
En prévention, il faut éviter les traumatismes et les traitements hormonaux. Un geste d’embolisation est parfois proposé par une équipe spécialisée.
Les malformations vasculaires décrites peuvent se combiner et s’associer à d’autres anomalies tissulaires. Le syndrome de Klippel-Trénaunay (malformation capillaro-veinolymphatique + hypercroissance du membre atteint) en est un exemple célèbre (
Remerciements au service de dermatologie de l’hôpital Necker pour l’iconographie.
1. Hémangiome infantile : place et apport des examens complémentaires ?
Ils n’ont d’intérêt qu’en cas de doute diagnostique , dans les formes sous-cutanées strictes, ou pour rechercher d’éventuelles anomalies associées ou complications.
L’écho-doppler montre une masse sous-cutanée bien limitée, d’échogénicité variable, avec une vascularisation mixte artérielle et veineuse. L’IRM met en évidence une masse lobulée aux limites nettes, iso-T1, hyper-T2, sans œdème périlésionnel, avec un rehaussement global précoce, homogène et prolongé.
Cependant, l’imagerie n’étant pas strictement spécifique, le diagnostic repose sur la confrontation radioclinique.
L’analyse histologique révèle une prolifération endothéliale bénigne, s’organisant en capillaires, et exprimant spécifiquement le GLUT-1 (transporteur de glucose de classe I) en immunohistochimie.
Risque vital (rare) :
• sous-glottique ;
• de grande taille : risque d’insuffisance cardiaque (hémangiome hépatique) ;
• cérébral ou médullaire (risque de compression) ;
• digestif (hémorragies).
Risque fonctionnel :
• orbitaire ou palpébral : risque d’amblyopie ;
• du conduit auditif ;
• labial ;
• nasal ;
• de la région périnéale à proximité d’un orifice.
Hémangiome ulcéré (risques d’infection, douleurs, saignement) :
• situé dans une zone de frottement (siège, lèvres, dos…), hémangiome segmentaire.
Risque esthétique :
• de la face de grande taille ;
• nodulaire du nez, des lèvres ou de la paupière ;
• sous-cutané du mamelon chez une fille.
3. Malformations veineuses multifocales
Syndrome de Bean
Sporadique. Malformations veineuses cutanées multiples de taille très variable ; atteinte digestive +++ avec risque hémorragique ; atteinte musculaire (douleurs) CIVL.
Syndrome de Maffucci
Sporadique. Malformations veineuses nodulaires, fermes, prédominant aux extrémités ; atteintes muqueuses ; enchondromes avec risque fracturaire et risque d’évolution vers un chondrosarcome +++ ; CIVL.
Malformations glomuveineuses familiales
Héréditaire (autosomique dominant). MV cutanées de taille variable, douloureuse à la pression et aux changements de température ; pas d’atteinte muqueuse ni CIVL
1. Wassef M, Blei F, Adams D, et al. Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 2015;136:e203-14.
2. Léauté-Labrèze C, Hoeger P, Mazereuw-Hautier J, et al. A randomized controlled trial of oral propranolol in infantile hemangioma. N Eng J Med 2015;372:735-46.
3. Boccara O, Puzenat E, Proust S, et al. The effects of sirolimus on Kasabach-Merritt Phenomenon coagulopathy. Br J Dermatol 2018; 178:e114-e116.
4. Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, et al. A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. J Am Acad Dermatol 2015;72:473-80.
5. Mazoyer E, Enjolras O, Bisdorff A, Wassef M, Drouet L. Coagulation disorders in patients with venous malformation of the limbs and trunk: a case series of 118 patients. Arch Dermatol 2008;144:861-7.
6. Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat 2013;34:1632-41.
7. Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet 2016; 172:402-21.
www.hemangiome.com
www.dermato-info.fr
www.maladiesrares-necker.aphp.fr/centre-de-reference-magec
Dans cet article
 Encadrés
Encadrés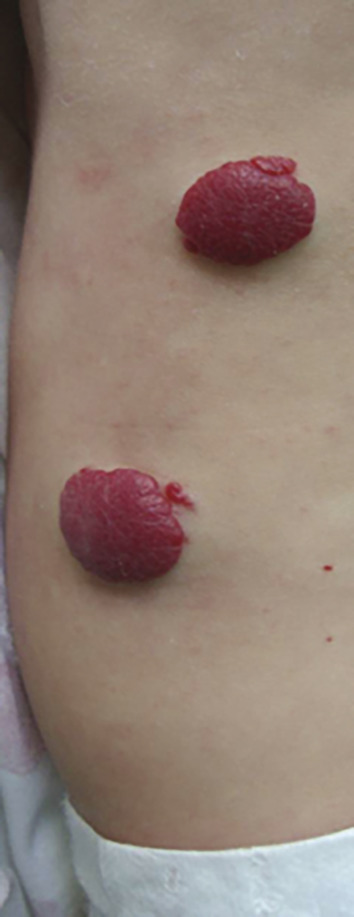
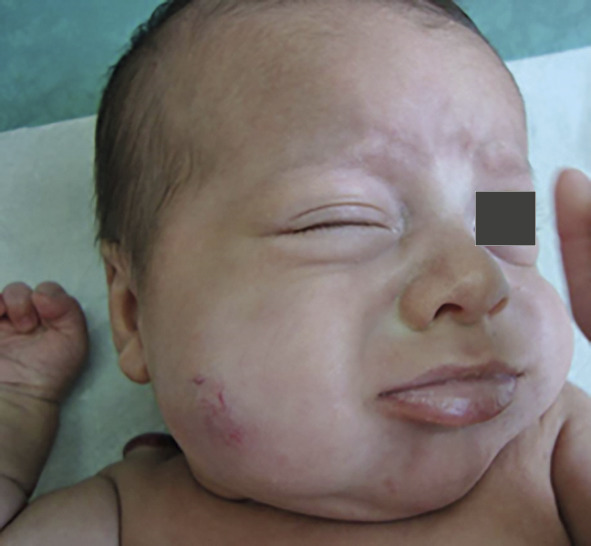
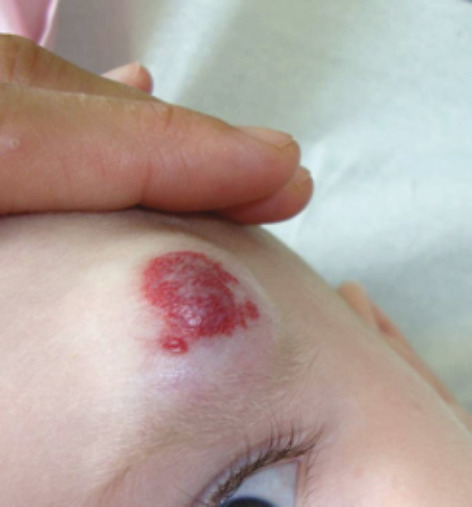
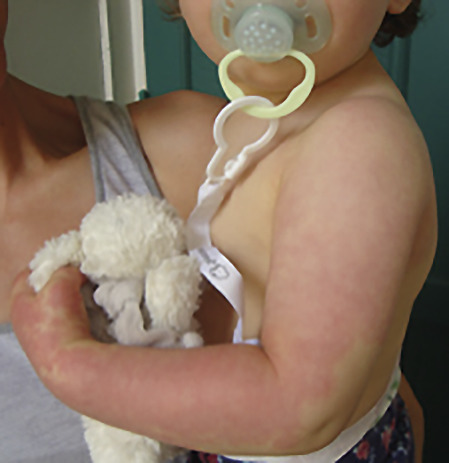
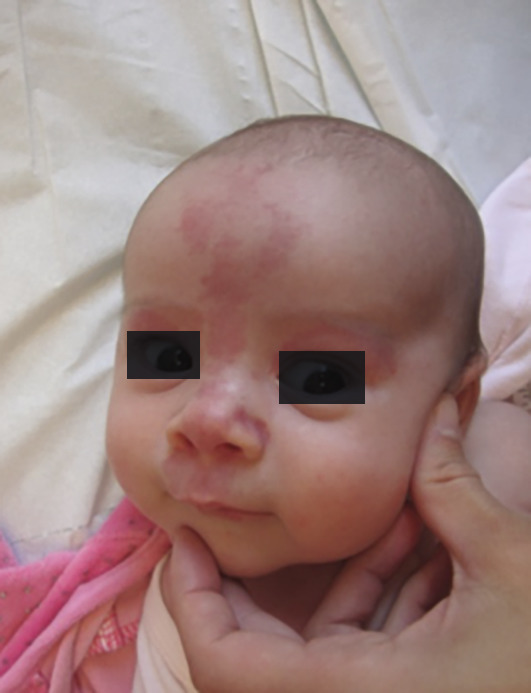
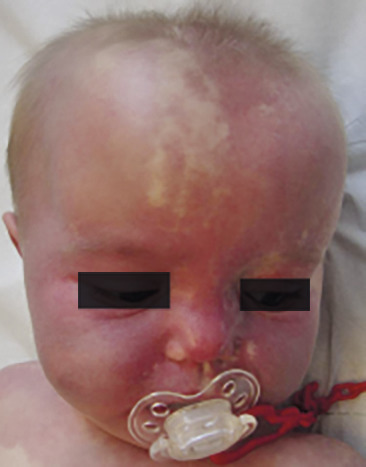
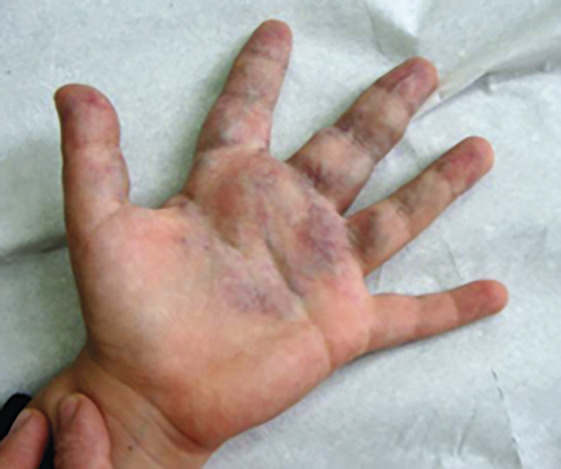

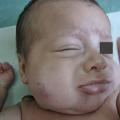
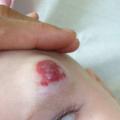
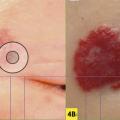
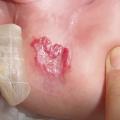
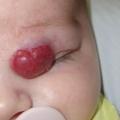
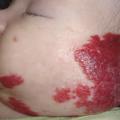
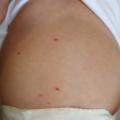
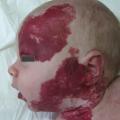
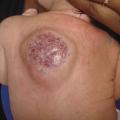
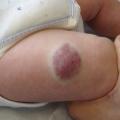
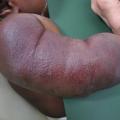
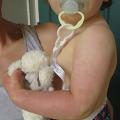

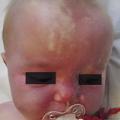
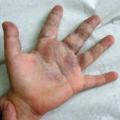
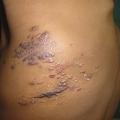
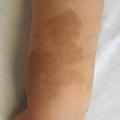
L’hémangiome infantile est l’atteinte vasculaire la plus fréquente chez les bébés.
Contrairement à l’angiome plan, c’est une lésion proliférative rapidement évolutive.
Rôle du MG : savoir reconnaître un hémangiome infantile requérant un traitement par propranolol pour l’adresser sans tarder à un centre expert.