La greffe de cellules souches hématopoïétiques autologues est déjà utilisée dans la prise en charge de certaines formes de sclérodermie systémique. L’injection de cellules stromales mésenchymateuses, actuellement à l’étude, promet d’élargir les indications de la thérapie cellulaire dans cette pathologie à la morbimortalité élevée.
La sclérodermie systémique (SSc) est une maladie auto-immune chronique rare marquée par une morbidité significative et une altération de la qualité de vie des patients atteints. La mortalité associée à la SSc est la plus élevée parmi toutes les maladies rhumatologiques, soulignant l’importance de développer de nouvelles approches thérapeutiques. La physiopathologie de la SSc repose sur une triade caractéristique, secondaire à une stimulation antigénique dans un contexte de susceptibilité génétique : atteinte endothéliale avec vasculopathie initiale, activation de la réponse immunitaire et, en conséquence, fibrose progressive de la peau et des différents organes. La thérapie cellulaire ouvre des perspectives, notamment par l’utilisation des cellules stromales mésenchymateuses (CSM), dont les propriétés immunomodulatrices, pro-angiogéniques et antifibrotiques sont bien caractérisées in vitro et in vivo. Les CSM peuvent être isolées à partir des nombreux tissus dans lesquels elles sont présentes : moelle osseuse, tissu adipeux, placenta, cordon ombilical et autres. Elles sont déjà utilisées en thérapie cellulaire avec des indications ciblées et validées dans certains pays : prévention du rejet de greffe de moelle, fistules de la maladie de Crohn. Elles sont actuellement à l’étude dans la sclérodermie systémique.
Sclérodermie systémique : une morbi-mortalité élevée
La sclérodermie systémique (SSc) est une maladie auto-immune rare et hétérogène. Sa prévalence varie de 7 à 1 580 par million et son incidence de 0,6 à 19 par million, avec une prédominance féminine (sex-ratio compris entre 3/1 et 14/1) entre 20 et 50 ans.
Le terme de sclérodermie (de skleros, dur, et derma, peau) fait référence à l’atteinte cutanée secondaire à l’augmentation de la production de collagène et à son accumulation dans la peau. Il existe également des formes sine scleroderma.
Triade pathogénique caractéristique
Les manifestations cliniques de la maladie découlent des 3 principaux mécanismes physiopathologiques qui, suite à une stimulation antigénique de nature variable dans un contexte de susceptibilité génétique, associent :
– une vasculopathie diffuse avec lésions endothéliales précoces des petits vaisseaux à l’origine du syndrome de Raynaud, d’ulcères digitaux, d’une hypertension artérielle pulmonaire ou d’une atteinte rénale (hypertension artérielle, crise rénale) ;
– une inflammation chronique avec des phénomènes d’auto-immunité (activation de la réponse immunitaire innée et adaptative), d’auto-inflammation (production de cytokines pro-inflammatoires) et apparition d’auto-anticorps spécifiques, qui sont des marqueurs diagnostiques et de classification de la maladie ;
– une fibrose progressive, plus ou moins extensive, touchant principalement la peau, le tube digestif, les poumons, le cœur et les reins.
D’importants besoins thérapeutiques
La sclérodermie systémique est associée à une morbidité élevée avec altération importante de la qualité de vie. Le taux de mortalité standardisé est multiplié d’un facteur 3,5 par rapport à la population générale. Les principales causes de décès sont cardiaques et pulmonaires, avec un taux de mortalité à 8-10 ans de 20-30 %. En cas d’évolution rapide et diffuse (10-20 % des patients), le taux de survie à 3-5 ans est estimé à 50-70 %, selon la nature et l’étendue des atteintes pulmonaire, cardiaque ou rénale, dont la présence est un facteur péjoratif. Ces données soulignent l’importance du traitement précoce en cas d’atteinte viscérale ou dans les formes cutanées rapidement progressives.1
Malgré les progrès diagnostiques et de prise en charge réalisés ces vingt dernières années, la sclérodermie systémique reste une maladie orpheline avec d’importants besoins thérapeutiques. Les traitements recommandés sont pour la plupart symptomatiques (contre le syndrome de Raynaud, le reflux gastro-œsophagien, l’hypertension artérielle) ; les médicaments immunomodulateurs (méthotrexate) ou immunosuppresseurs classiques (cyclophosphamide, mycophénolate mofétil), utilisés dans les formes précoces sévères ou rapidement évolutives, ont des effets modestes et n’améliorent pas la survie.1
Plusieurs nouvelles voies thérapeutiques sont à l’étude : immunothérapies ciblées sur la signalisation des cytokines (interleukine 6 [IL-6], par exemple), les lymphocytes B avec thérapie anti-CD20, les lymphocytes T avec inhibition de leur costimulation, ou d’autres voies de signalisation spécifiques de la fibrose (transforming growth factor [TGF-β]). En mars 2021, le tocizulimab (antagoniste du récepteur à l’IL-6) a été approuvé par la Food and Drug Administration aux États-Unis pour les atteintes pulmonaires interstitielles dans la sclérodermie systémique ; le nintédanib (inhibiteur de tyrosine kinase [TKi]) a également été autorisé dans cette même indication aux États-Unis et en Europe, à visée antifibrosante.
L’espoir de la thérapie cellulaire : succès de la greffe de cellules souches autologues
Dans ce contexte, l’utilisation d’une nouvelle approche, basée sur la thérapie cellulaire, ouvre des espoirs thérapeutiques chez les patients atteints de sclérodermie systémique. Ainsi, l’administration d’une chimiothérapie intensive suivie d’une greffe de cellules souches hématopoïétiques (CSH) autologues (préalablement prélevées ou mobilisées à partir de la moelle osseuse), a fait progressivement depuis 20 ans la preuve de son efficacité pour traiter les formes sévères (qui représentent 20 à 30 % de la population des patients). Cette technique est aujourd’hui validée par toutes les sociétés savantes avec un niveau de preuve de grade 1 en France et à l’international. Pour la première fois dans cette affection, l’utilisation de l’autogreffe de CSH a permis d’obtenir une régression effective de la fibrose cutanée et pulmonaire avec amélioration de la survie globale et de la survie sans événement jusqu’à 5-7 ans.
Cependant, l’utilisation de cette procédure reste réservée aux patients les plus atteints, et est contre-indiquée à un stade trop tardif de la maladie. La greffe de cellules stromales mésenchymateuses élargit alors le champ des possibles.
Cellules stromales mésenchymateuses : des atouts certains
Les cellules stromales mésenchymateuses (CSM), identifiées pour la première fois dans la moelle osseuse par Friedenstein en 1976,2 sont désormais bien caractérisées ; elles peuvent être produites à partir de tissu adipeux, de cordon ombilical ou de gelée de Wharton, et de nombreux autres tissus. Ces cellules progénitrices multipotentes ont des propriétés immunomodulatrices et immunosuppressives, pro-angiogéniques et antifibrotiques, justifiant leur utilisation pour cibler les différents effecteurs de la triade pathogénique au cours de la SSc.
Des cellules multipotentes…
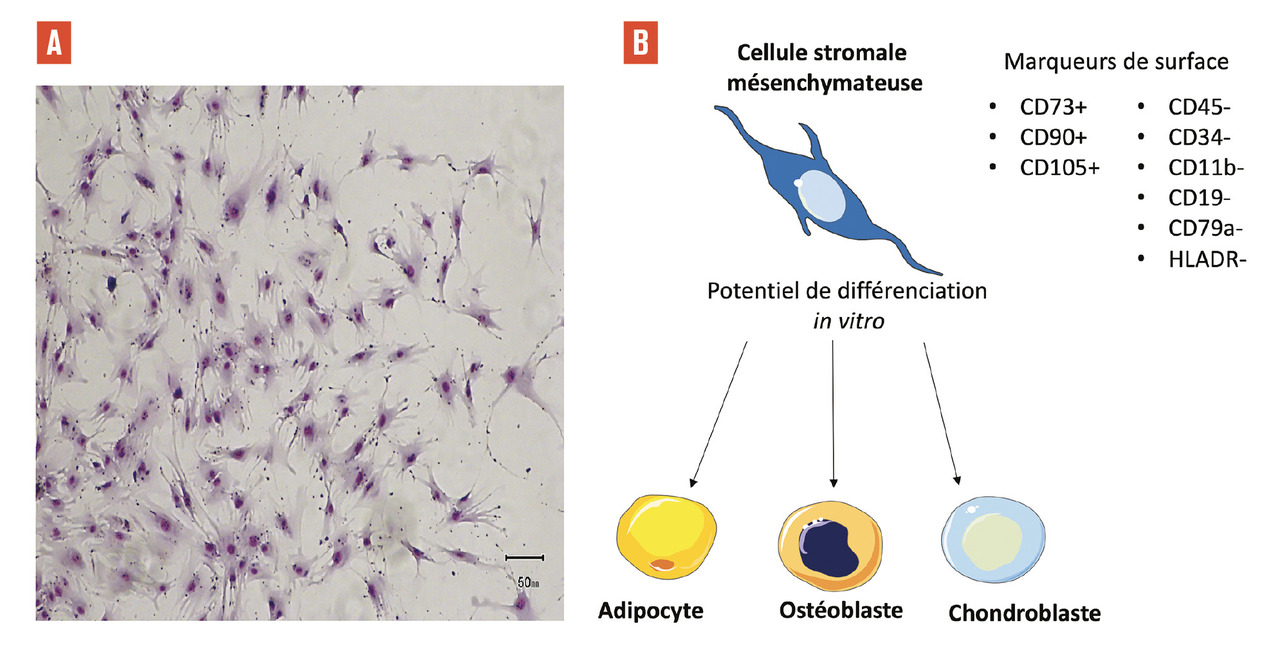
Les CSM, initialement décrites comme les précurseurs des cellules osseuses, ressemblent à des fibroblastes et sont capables de se différencier dans la voie ostéogénique. Elles font partie du micro-environnement médullaire hautement spécialisé, au sein duquel elles participent notamment à la régulation de l’autorenouvellement et de la différenciation des cellules souches hématopoïétiques. Des études in vitro ont montré que les CSM sont capables de se différencier vers d’autres lignées cellulaires mésodermiques : chondrocytes, adipocytes et myoblastes.
En 1991, Caplan a introduit le terme de « cellules souches mésenchymateuses »,3 qui fut ensuite plus exactement modifié en « cellules stromales multipotentes », puisque la définition d’une cellule souche repose avant tout sur sa capacité d’autorenouvellement in vitro et in vivo avec différenciation clonale possible en différentes lignées cellulaires. Aussi, les CSM sont des « cellules stromales multipotentes », plus récemment décrites par Caplan lui-même comme des « cellules stromales médicinales » compte tenu de leur mode d’action cellulaire au site de l’inflammation.
… dont les critères phénotypiques sont bien identifiés
Selon des critères établis a minima, en 2006, par la Société internationale de thérapie cellulaire (ISCT),4 les CSM sont une population cellulaire définie in vitro par les caractéristiques phénotypiques suivantes :
– polyclonale, adhérente au plastique, avec une morphologie de type fibro-blaste ;
– exprimant les antigènes de surface CD73+, CD90+, CD105+ (pour plus de 95 % des CSM) ;
– n’exprimant pas les marqueurs hématopoïétiques et endothéliaux (CD45-, CD34-, CD14-, CD11b-, CD19-, CD79a- et HLADR-) ;
– capable de se différencier in vitro en ostéoblastes, adipocytes et chondroblastes (
Études in vitro justifiant leur intérêt dans la sclérodermie systémique
La plupart des études in vitro justifiant l’utilisation des CSM dans la sclérodermie systémique s’intéressent à la description de leurs propriétés immunorégulatrices.5, 6
Propriétés multiples prometteuses
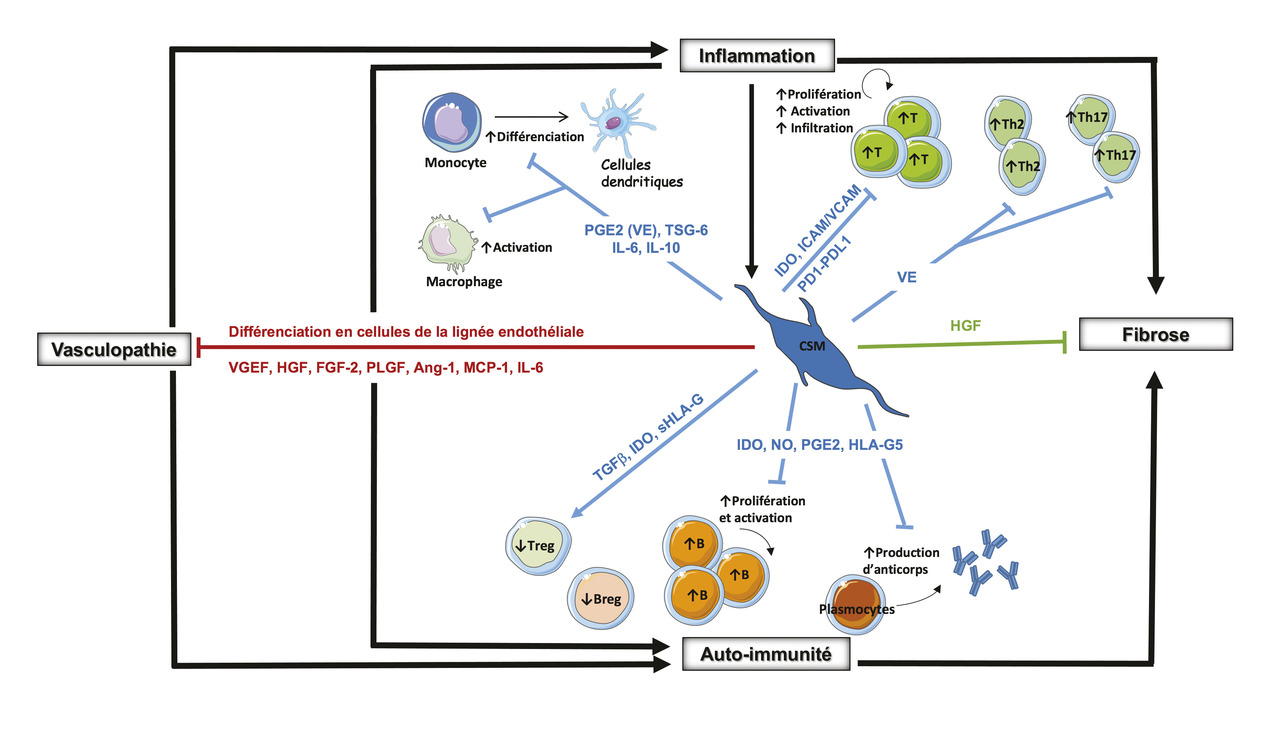
Les CSM ont montré leur capacité à inhiber in vitro la prolifération et l’activation excessive des lymphocytes T et B au cours de la maladie.
Par ailleurs, les CSM agissent au niveau du système immunitaire inné, et leur utilisation concourt également à augmenter le nombre et l’activation des lymphocytes T régulateurs (Treg) et des lymphocytes B régulateurs (Breg) [
Les propriétés proangiogéniques des CSM sont démontrées par leurs capacités à se différencier en cellules de la lignée endothéliale et à favoriser la formation de nouveaux vaisseaux sanguins.
Enfin, leur capacité à inhiber la prolifération de fibroblastes pulmonaires et à améliorer la réparation de tissu épithélial pulmonaire confirme leurs propriétés antifibrotiques.
Modes d’action : plutôt par sécrétion de facteurs solubles
Certains effets des CSM sont médiés par contact direct de cellule à cellule.
La majeure partie de leur action s’exerce néanmoins par sécrétion des différents facteurs solubles, qui ne sont pas constitutivement exprimés par les CSM, mais sont induits par les stimuli inflammatoires présents dans le milieu local (notamment en présence d’interféron gamma [IFN-γ], voire des tumor necrosis factor alpha [TNF-α], interleukines alpha ou bêta [IL-1α ou IL-1β]). En effet, ces stimuli permettent la production de facteurs solubles par les CSM : des facteurs de croissance (transforming growth factor beta [TGF-β], hepatocyte growth factor [HGF], vascular endothelial growth factor [VEGF], angiopoïétine-1 [Ang-1]), des cytokines (prostaglandine E2, IL-6, IL-10), la molécule HLA-G ou encore l’enzyme métabolique indoléamine 2,3-dioxygénase (IDO), marqueur d’activité des CSM (
Tous ces facteurs contribuent aux activités anti-inflammatoire, immunomodulatrice, proangiogénique et antifibrosante des CSM démontrées in vitro et in vivo dans différentes indications : greffe de moelle, maladies auto-immunes rhumatologiques, neurologiques, gastroentérologiques, vasculaires.
Source des CSM : un facteur d’hétérogénéité
Les sources les plus fréquentes de CSM à usage thérapeutique sont la moelle osseuse, le tissu adipeux, le cordon ombilical, la gelée de Wharton contenue dans le cordon ombilical et le placenta.
Les CSM issues de ces tissus partagent de nombreuses caractéristiques biologiques, mais diffèrent dans leur potentiel de prolifération, leur profil transcriptionnel global et de fonctionnalité.
Ainsi les CSM issues de la moelle osseuse (CSM-MO) ont des capacités de prolifération et de différenciation qui diminuent avec l’âge et varient considérablement selon les donneurs. Le prélèvement de moelle est une procédure invasive pour le donneur, avec un risque d’exposition virale chez le receveur.
Les CSM d’origine adipeuse (CSM-TA) sont isolées à partir de la fraction vasculaire stromale du tissu adipeux sous-cutané qui peut produire jusqu’à 500 fois plus de CSM que la moelle osseuse.
Récemment, les premières analyses transcriptomiques, phénotypiques et fonctionnelles sur des échantillons appariés de CSM-MO et CSM-TA provenant du même donneur ont montré une plus forte inhibition des réponses immunitaires par CSM-TA et une immunogénicité plus faible.7
In vivo, dans différents modèles murins de sclérodermie systémique, l’action anti-inflammatoire et antifibrosante des CSM issues du tissu adipeux, ombilical ou de la moelle osseuse a été confirmée à des degrés divers après leur injection.5, 6
In vitro comme in vivo, d’autres paramètres modulent les capacités fonctionnelles des CSM : notamment les caractéristiques du donneur, le processus de production et les conditions de culture, l’administration au patient de CSM cryoconservées puis décongelées ou issues de cultures « fraîches », et l’environnement inflammatoire du receveur. Ces notions sont essentielles pour l’interprétation des résultats observés dans les études, de même que l’identification de marqueurs permettant de prédire l’efficacité attendue en clinique humaine.
Utilisation des CSM à visée thérapeutique chez l’homme
Les propriétés immunosuppressives, immunomodulatrices et trophiques des CSM sont donc bien démontrées in vitro et chez l’animal.
Des études pour des indications multiples
La mise en place progressive d’études pilotes puis randomisées chez l’homme permet d’évaluer leur efficacité thérapeutique dans différentes indications : tout d’abord après greffe de moelle dans le traitement de la maladie du greffon contre l’hôte, puis dans la prévention du rejet de greffe (rénale et hépatique), dans différentes maladies auto-immunes et auto-inflammatoires (lupus érythémateux systémique, syndrome de Gougerot-Sjögren, maladie de Crohn, sclérose en plaques), en neurologie et en médecine vasculaire (prévention ou traitement de l’ischémie aiguë cérébrale et vasculaire périphérique).
En dépit des nombreux essais cliniques menés, très peu ont conduit à la mise sur le marché de CSM à visée thérapeutique. En mars 2022, la base de données ClinicalTrials.gov rapportait 151 essais thérapeutiques à l’échelon mondial analysant l’injection de CSM autologues ou allogéniques à partir de donneurs sains dans les maladies auto-immunes, dont 6 utilisaient des CSM allogéniques dans la SSc (
CSM allogéniques dans le traitement de la sclérodermie systémique
Plusieurs travaux menés en parallèle il y a dix ans ont permis de mettre en évidence in vitro et in vivo l’existence d’anomalies constitutives des CSM présentes chez les patients atteints de maladies auto-immunes, notamment la sclérodermie8 mais aussi le lupus et la polyarthrite rhumatoïde. Ces résultats acquis ont souligné l’importance d’utiliser des CSM d’origine allogénique, prélevées chez des donneurs sains pour une fonctionnalité optimale.
Efficacité et bonne tolérance
L’utilisation des CMS allogéniques, issues de la moelle osseuse ou du cordon ombilical, dans le traitement de la SSc sévère a été initialement rapportée en Europe chez 6 patients, avec un suivi clinique après injection de 6 et 44 mois (
Une étude chinoise a ensuite étudié l’association de la combinaison des plasmaphérèses et du cyclophosphamide avec l’injection de CSM allogéniques chez 14 patients avec SSc, rapportant également une amélioration globale du mRSS, ainsi que l’augmentation des paramètres de la fonction pulmonaire chez 3 patients.11
Vers des biomarqueurs d’efficacité thérapeutique spécifiques
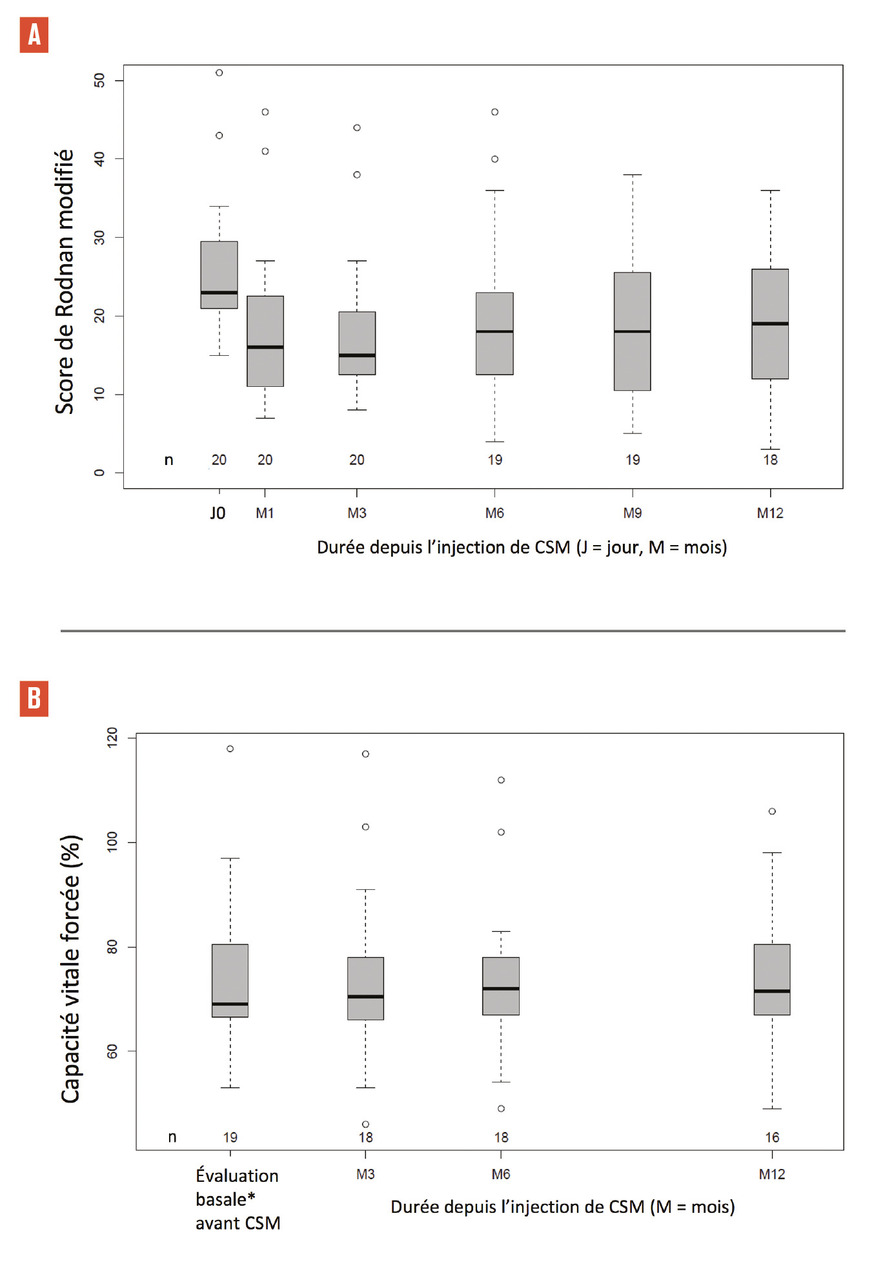
Depuis lors, notre équipe a réalisé la première étude clinique de phase I-II ouverte, non randomisée, monocentrique, analysant l’effet de doses croissantes de CSM allogéniques issues de moelle osseuse chez 20 patients atteints de SSc sévères et réfractaires aux traitements préalables.12 Vingt et un donneurs intrafamiliaux sains différents ont été nécessaires en raison d’un échec de la culture des CSM chez un donneur. Le protocole prévoyait d’administrer la dose de 1.106 CSM/kg aux 10 premiers patients et 3.106 CSM/kg aux 10 patients suivants en cas de faible probabilité de toxicité à 1.106 CSM/kg, ce qui a été objectivé à l’analyse intermédiaire de l’étude. Après un suivi médian de 24,1 mois (écart interquartile [IQR] : 20,8-24,5), les résultats cliniques observés chez les patients traités pour SSc ont démontré l’innocuité (objectif principal) d’une seule perfusion de CSM-MO allogéniques (1 à 3.106 cellules/kg). Une diminution précoce de la fibrose cutanée (objectivée par le mRSS) a également été observée trois à six mois après l’injection et a persisté jusqu’à douze mois de suivi. Par ailleurs, les paramètres des fonctions pulmonaire (
Durée d’exposition aux csm, facteur d’efficacité majeur
La plupart des études humaines testant l’injection de CSM dans le traitement de maladies auto-immunes ont utilisé une dose de cellules de 1 à 5 millions de CSM/kg, sans différence d’efficacité selon les doses utilisées. Compte tenu de la biodistribution des CSM (disparition rapide après perfusion intraveineuse) et de leur mécanisme d’action (facteurs sécrétés), le concept de perfusions répétées gagne du terrain dans le domaine. Les données suggèrent en effet que la durée d’exposition aux CSM est probablement plus importante que l’intensité de l’exposition. Ces résultats très encourageants ouvrent la voie à d’autres études contrôlées randomisées conçues pour évaluer l’innocuité et l’efficacité des injections répétées de CSM allogéniques provenant de différentes sources de tissus pour traiter les patients atteints de SSc (
1. Hachulla E, Agard C, Allanore Y, Avouac J, Bader-Meunier B, Belot A, et al. Protocole national de diagnostic et de soins sur la sclérodermie systémique. Actualisation 2020. Haute Autorité de santé 2017;140.
2. Friedenstein AJ, Gorskaja JF, Kulagina NN. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Hematol 1976;4(5):267-74.
3. Caplan AI. Mesenchymal stem cells. J Orthop Res 1991;9(5):641-50.
4. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8(4):315-7.
5. Farge D, Loisel S, Lansiaux P, Tarte K. Mesenchymal stromal cells for systemic sclerosis treatment. Autoimmun Rev 2021;102755.
6. Farge D. Mesenchymal stromal cells for systemic sclerosis. In: Hematopoietic stem cell transplantation and cellular therapies for autoimmune diseases. CRC Press; 2021.
7. Menard C, Dulong J, Roulois D, Hebraud B, Verdiere L, Pangault C, et al. Integrated transcriptomic, phenotypic, and functional study reveals tissue-specific immune properties of mesenchymal stromal cells. Stem Cells 2020;38(1):146-59.
8. Vanneaux V, Farge-Bancel D, Lecourt S, Baraut J, Cras A, Jean-Louis F, et al. Expression of transforming growth factor beta receptor II in mesenchymal stem cells from systemic sclerosis patients. BMJ Open [Internet] 2013;3(1). Consultable sur http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23299111
9. Christopeit M, Schendel M, Foll J, Muller LP, Keysser G, Behre G. Marked improvement of severe progressive systemic sclerosis after transplantation of mesenchymal stem cells from an allogeneic haploidentical-related donor mediated by ligation of CD137L. Leukemia 2008;22(5):1062-4.
10. Keyszer G, Christopeit M, Fick S, Schendel M, Taute BM, Behre G, et al. Treatment of severe progressive systemic sclerosis with transplantation of mesenchymal stromal cells from allogeneic related donors: report of five cases. Arthritis Rheum 2011;63(8):2540-2.
11. Zhang H, Liang J, Tang X, Wang D, Feng X, Wang F, et al. Sustained benefit from combined plasmapheresis and allogeneic mesenchymal stem cells transplantation therapy in systemic sclerosis. Arthritis Research & Therapy 2017;19(1):165.
12. Farge D, Loisel S, Resche-Rigon M, Lansiaux P, Colmegna I, Langlais D, et al. Safety and preliminary efficacy of allogeneic bone marrow-derived multipotent mesenchymal stromal cells for systemic sclerosis: a single-centre, open-label, dose-escalation, proof-of-concept, phase 1/2 study. The Lancet Rheumatology 2022;S266599132100326X.