La mucoviscidose est due à des mutations du gène CFTR. Depuis dix ans, des molécules capables de restaurer la fonction de la protéine CFTR ont été développées, offrant une nouvelle voie thérapeutique à une grande majorité de patients.
La mucoviscidose est la maladie génétique mortelle la plus fréquente dans la population occidentale, touchant 1 individu pour 3 500 naissances en Europe. Dans le monde, 70 000 patients sont atteints de mucoviscidose, et ils sont 7 300 en France.1 La mucoviscidose est une maladie à transmission autosomale récessive, due à des mutations du gène CFTR (Cystic Fibrosis Transmembrane conductance Regulator) qui code pour la protéine du même nom. La protéine CFTR est exprimée au niveau de multiples épithéliums, ce qui explique les complications multiviscérales de la maladie, avec une atteinte prépondérante du poumon et du pancréas. L’identification du gène CFTR et de sa mutation la plus fréquente F508del (Phe508del) date de 1989. À la même période, l’organisation des soins s’est structurée : meilleure prise en charge respiratoire (développement des techniques de kinésithérapie respiratoire particulières, apparition des molécules inhalées spécifiques de la mucoviscidose), anti-infectieuse et nutritionnelle, avec l’introduction des enzymes pancréatiques gastroprotégées. L’organisation des centres de soins s’est complétée en France en 2002 avec la labélisation des centres de ressources et de compétences de la mucoviscidose (CRCM) concomitamment au déploiement du dépistage néonatal généralisé de la maladie, permettant d’améliorer la prise en charge précoce et le suivi des enfants.
L’amélioration organisationnelle et thérapeutique, l’identification du gène et de ses nombreux variants, la découverte de leur impact sur la synthèse de la protéine CFTR et sur l’expression phénotypique de la maladie ont été un apport considérable pour avancer dans la compréhension des fonctions de la protéine, de la physiopathologie de la maladie, dans l’aide au diagnostic des formes complètes ou incomplètes et, aujourd’hui, dans l’accès à des traitements personnalisés.2, 3 Ainsi, depuis dix ans, des molécules capables de restaurer la fonction de la protéine CFTR ont été développées, offrant actuellement une nouvelle voie thérapeutique à la grande majorité des malades.
Atteinte bronchopulmonaire : premier facteur pronostique
Le registre français de la mucoviscidose de 2020 a recensé 7 376 patients, parmi lesquels 60 % ont plus de 18 ans.1 83 % des patients sont porteurs de la mutation F508del, dont 41,3 % sont homozygotes et 41,4 % sont hétérozygotes composites, porteurs d’une mutation F508del et d’une autre mutation.
L’atteinte bronchopulmonaire de la mucoviscidose représente la cause la plus importante de morbi-mortalité de la maladie. Les traitements à visée respiratoire sont primordiaux et étaient essentiellement symptomatiques jusqu’à l’avènement des modulateurs de CFTR. Le déclin de la fonction respiratoire, mesuré par le volume expiratoire maximal par seconde (VEMS), est estimé à 1 % par an et il est impacté par la survenue d’exacerbations pulmonaires. Ces deux éléments marquent le pronostic de la maladie et sa morbi-mortalité.4, 5
Même si le nombre de décès à l’âge pédiatrique est aujourd’hui faible, la mucoviscidose reste une maladie mortelle ; l’âge médian au décès était de 32 ans en 2020 contre 27 ans en 2010. Le taux brut de mortalité diminue régulièrement depuis une décennie, passant de 10,3 à 6,2.1 55 % des décès concernent aujourd’hui des patients transplantés, qui représentent environ 20 % de la population adulte.
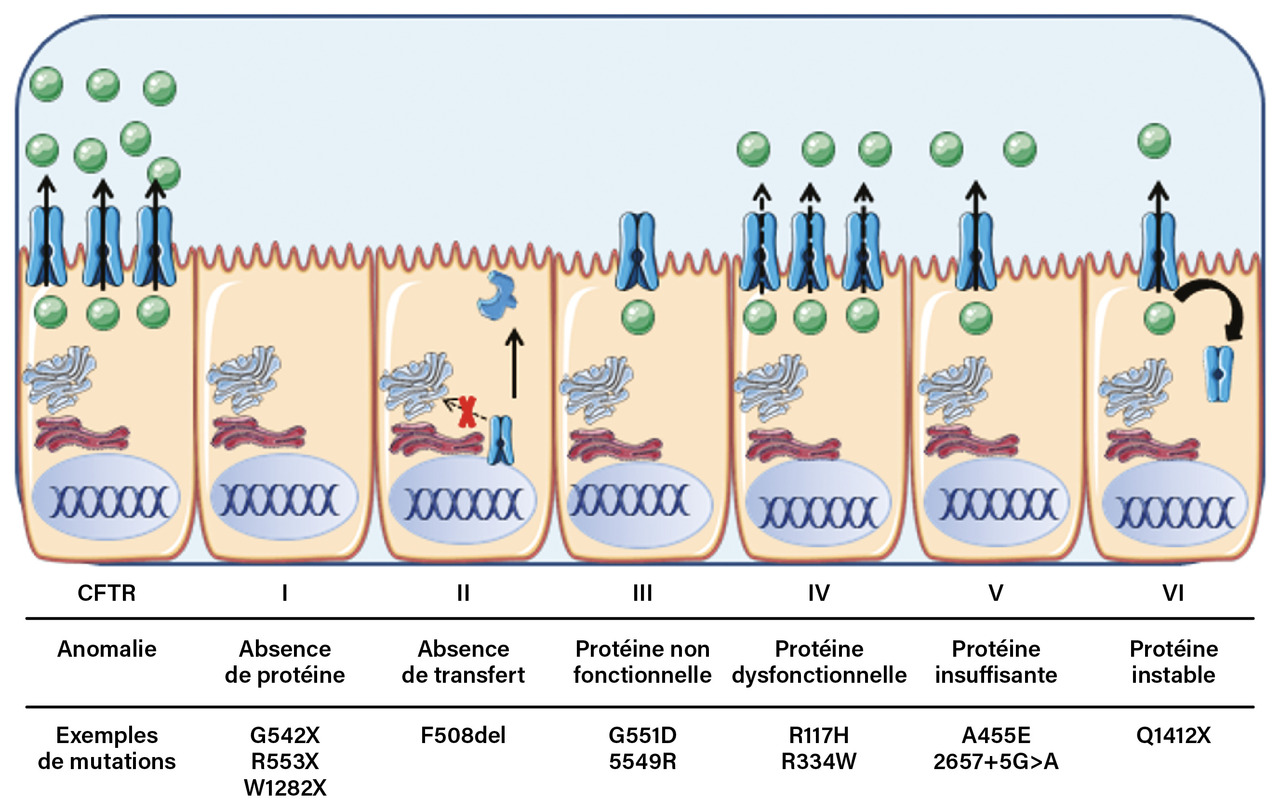
Six classes de mutations du gène CFTR
La mucoviscidose se transmet sur le mode autosomal récessif. Elle est due à des mutations dans le gène CFTR, localisé sur le bras long du chromosome 7 en position 7q31.2, qui code la protéine CFTR, protéine membranaire ATP-dépendante située au pôle apical de l’ensemble des cellules épithéliales de l’organisme (voies aériennes, intestin, pancréas exocrine, voies biliaires, tractus génital, glandes sudorales). Sa principale fonction est celle d’un canal ionique qui provoque une sortie active des ions chlorures (Cl-) et bicarbonates (HCO3-) de la cellule vers la lumière bronchique entraînant un mouvement d’eau et d’ions sodium (Na+) dans le même sens, assurant une hydratation correcte du liquide périciliaire ; la protéine CFTR régule également d’autres canaux présents à la surface cellulaire dont le canal sodium ENaC qui, lui, provoque une entrée active de Na+ dans la cellule. En l’inhibant, la protéine CFTR entraîne une amplification de son action d’hydratation du liquide périciliaire. Les mutations du gène CFTR sont à l’origine d’un défaut quantitatif et/ou fonctionnel de la protéine CFTR.
Plus de 2 000 variants du gène CFTR sont connus. Ces mutations sont classées en six catégories (
Les mutations de classe I sont des mutations non-sens ; elles représentent 10 % environ des mutations de CFTR dans le monde, même si elles sont plus fréquentes dans la population israélienne où 60 % des patients sont porteurs de cette classe de mutation.
La délétion de la phénylalanine en position 508 (F508del) est responsable des anomalies de migration au pôle apical des cellules de la protéine mutée et d’une dégradation prématurée dans le système réticulo-endoplasmique (mutation de classe II).
Les mutations de classe III, appelées aussi « gating mutations », ne concernent qu’un petit pourcentage de patients (5 %). Parmi celles-ci, la plus fréquente est la mutation G551D. Les patients qui en sont porteurs ont une atteinte respiratoire et digestive classique. C’est pour les patients porteurs de cette mutation que le premier modulateur de la protéine CFTR, le VX-770 (ivacaftor [Kalydeco]) a obtenu une autorisation de mise sur le marché (AMM) en novembre 2012.6
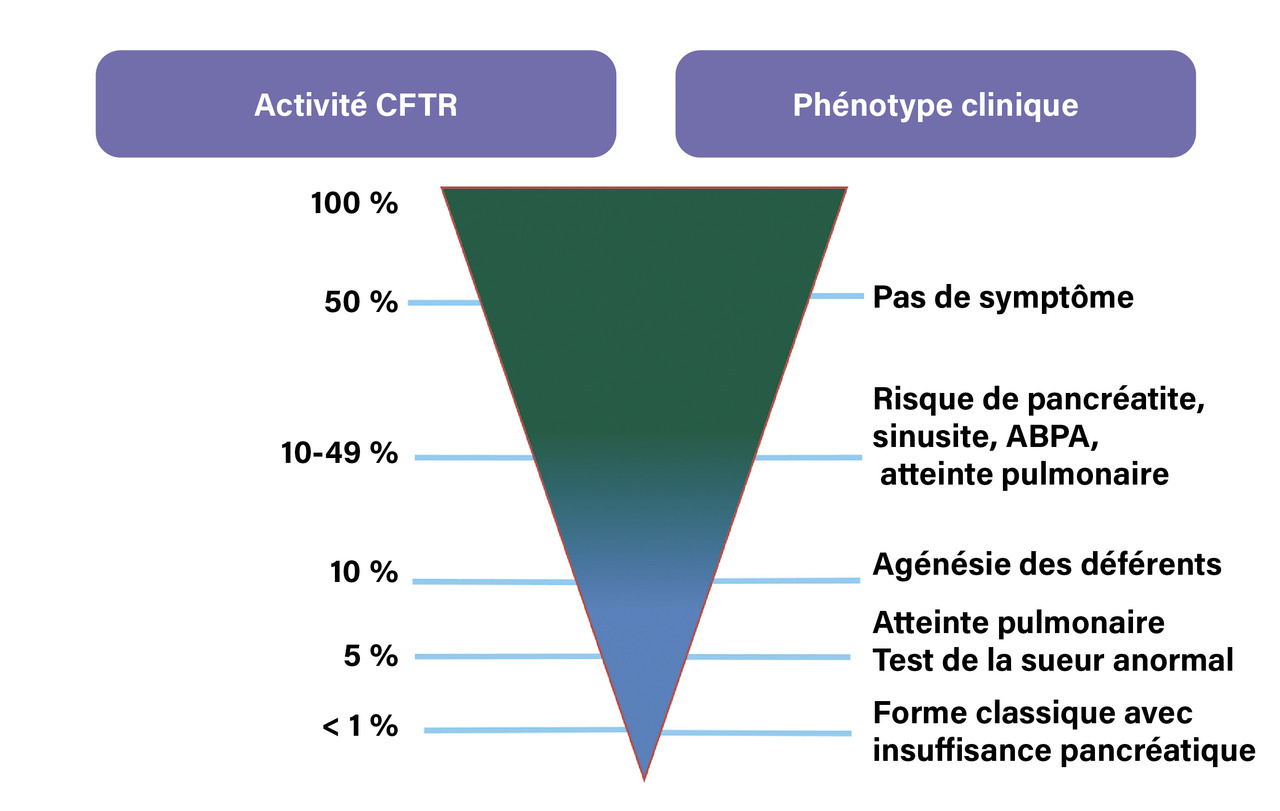
Les mutations de classes IV, V et VI sont généralement responsables de formes plus modérées de la maladie car associées à la présence d’une protéine partiellement fonctionnelle. La corrélation entre le génotype et le phénotype clinique est complexe et globalement peu prévisible d’emblée sauf pour le pancréas, dont la fonction exocrine est atteinte très précocement et de façon définitive dans les formes classiques de la maladie (
Modulateurs du canal CFTR : une révolution thérapeutique
Depuis dix ans, des potentiateurs et correcteurs de la fonction protéique CFTR ont été développés ; ils représentent une révolution thérapeutique pour la plupart des patients.
Ivacaftor, un potentiateur du canal CFTR
Les mutations dites gating représentent en France moins de 5 % des mutations. Elles altèrent l’ouverture du canal ionique CFTR.
En 2011, un essai clinique randomisé sur ivacaftor (Kalydeco) contre placebo en double aveugle montrait, chez des patients de plus de 12 ans porteurs de la mutation G551D, une amélioration significative de la fonction pulmonaire (augmentation de 10 points du VEMS), une diminution des surinfections pulmonaires de 55 %, et permettait une reprise pondérale de 3 kg.6 L’ivacaftor améliorait très significativement le score de qualité de vie.
Deux ans plus tard, une étude comparable menée chez des enfants porteurs de cette même mutation G551D confortait les données de l’étude princeps et montrait pour la première fois la normalisation de la concentration de chlorure sudoral (inférieure à 60 mmol/L), témoignant de l’effet systémique du médicament.7 Les études ultérieures ont permis d’élargir l’indication à d’autres mutations de la même classe et aux nourrissons de 4 mois et plus avec même, chez certains d’entre eux, la préservation de la fonction pancréatique.
Association d’un potentiateur et d’un correcteur du canal CFTR en cas de mutation F508del
Plus de 80 % des mutations en France sont de type F508del et sont caractérisées par une protéine CFTR anormalement conformée qui est en grande partie détruite par le protéasome cellulaire. La première association testée a été lumacaftor-ivacaftor (Orkambi) dans deux essais cliniques randomisés contre placebo en double aveugle chez des patients de plus de 12 ans homozygotes pour la mutation CFTR F508del.8 Même si l’amélioration de la fonction respiratoire était moindre (gain de VEMS de 2,6 à 4 points), l’efficacité sur la diminution des exacerbations pulmonaires ainsi que sur le nombre de cures d’antibiotiques et d’hospitalisations était maintenue. L’AMM et le remboursement de cette association ont été obtenus en France en décembre 2015 pour les patients de 12 ans et plus, et élargi aux enfants de plus de 2 ans depuis décembre 2019. Une bithérapie de deuxième génération par tezacaftor-ivacaftor (Symkevi) a été évaluée dans deux essais cliniques de phase III randomisés contre placebo en double insu, le premier chez des patients âgés de plus de 12 ans homozygotes pour F508del,9 le deuxième chez des hétérozygotes composites F508del/mutation résiduelle.10 Le VEMS augmentait de 6,8 points, et le taux d’exacerbations pulmonaires diminuait de 35 %. Le remboursement ayant été obtenu tardivement en France, il n’a été que peu prescrit.
Triple association d’un potentiateur et de deux correcteurs : remarquablement efficace
Plusieurs essais ont été successivement menés, concluant tous à une efficacité inédite de la triple association chez les patients porteurs d’au moins une mutation F508del.
Premier essai concluant
Le premier essai mené dans 115 centres dans 13 pays entre 2018 et 2019 a randomisé des patients hétérozygotes pour la mutation F508del entre traitement associant elexacaftor-tezacaftor-ivacaftor, et placebo.11 La triple association a permis d’améliorer le VEMS de 13,8 points dès quatre semaines de traitement ; le taux d’exacerbations pulmonaires a diminué de 63 % ; l’amélioration de la qualité de vie a été très significative et la concentration en chlorure sudoral a diminué de 41,8 mmol/L.
Deuxième étude : contre bithérapie
Le deuxième essai mené dans 44 centres de 4 pays en Europe entre août et décembre 2018 a randomisé des patients âgés de plus de 12 ans homozygotes pour la mutation F508del entre l’association elexacaftor-tezacaftor-ivacaftor et un traitement contrôle actif (tezacaftor-ivacaftor).12 Le VEMS a été significativement amélioré de 10 points, la concentration en chlorure sudoral a diminué de 45 mmol/L, revenant à des valeurs normales.
Troisième essai confirmant les deux autres
Le troisième essai mené dans 96 centres en Amérique du Nord, Europe et Australie entre août 2019 et juin 2020 a randomisé des patients âgés de plus de 12 ans avec un génotype F508del/fonction résiduelle entre l’association elexacaftor-tezacaftor-ivacaftor et un traitement contrôle actif par ivacaftor seul ou tezacaftor-ivacaftor.13 La triple association a obtenu une amélioration du VEMS et une réduction de la concentration en chlorure sudoral.
Amélioration de la qualité de vie
Très récemment a été publié un nouvel essai clinique multicentrique (35 centres dans 4 pays) randomisé comparant la triple association elexacaftor-tezacaftor-ivacaftor à un comparateur actif associant tezacaftor-ivacaftor chez des patients âgés de plus de 12 ans homozygotes pour la mutation F508del.14 Pour la première fois, le critère de jugement principal reposait sur l’amélioration de la qualité de vie et non pas sur celle du VEMS ; les résultats étaient significatifs.
Un bouleversement thérapeutique !
Les modulateurs du canal CFTR ont été valorisés, avec la reconnaissance d’une amélioration du service médical rendu (ASMR) de niveau II (important) pour l’ivacaftor (Kalydeco) et pour la combinaison ivacaftor-tezacaftor-elexacaftor (Kaftrio). La triple association est disponible en France depuis décembre 2019 en procédure d’accès précoce pour les patients sévères et elle est maintenant remboursée pour tous les patients homozygotes ou hétérozygotes F508del de plus de 12 ans. Après les déceptions liées aux résultats des essais de thérapie génique in vivo au début des années 2000, l’arrivée de la trithérapie modulatrice du CFTR a constitué un formidable progrès pour les patients atteints de mucoviscidose.
L’efficacité et la tolérance de l’ivacaftor ou de la combinaison ivacaftor-tezacaftor-elexacaftor devraient bouleverser l’évolution de la maladie pour les patients concernés. Leur impact sur la qualité de vie est d’ores et déjà tangible par la réduction spectaculaire des symptômes, notamment respiratoires.15 Les parcours de soins des patients traités par ces nouveaux traitements vont devoir faire l’objet de travaux ciblés, mais il apparaît déjà que le temps quotidien consacré aux traitements symptomatiques est allégé. Et une baisse très importante du recours à la transplantation pulmonaire a d’ores et déjà été observée.
Le traitement par Kaftrio a pu être initié chez plus de 2 000 patients fin 2021, et environ 5 000 patients devraient pouvoir en bénéficier en 2022 lorsque les jeunes enfants pourront se le voir prescrire.
Quelques limites : données à préciser et patients en attente
Il reste maintenant à démontrer l’efficacité de ces nouvelles thérapeutiques à long terme sur l’espérance de vie, ainsi que leur tolérance, notamment hépatique, après de longues durées d’exposition puisqu’il s’agit d’un traitement à vie. Son initiation devrait être possible dès le plus jeune âge (extensions d’AMM en cours).
15 % des patients sont porteurs de mutations actuellement non éligibles à l’indication de la triple thérapie, mais certaines de ces mutations à fonction minimale ou résiduelle semblent répondre in vitro. Elles font donc l’objet d’essais cliniques, voire d’accès précoce, pour les patients avec atteinte respiratoire sévère.
Pour les patients porteurs de mutations de classe I (mutations « stop » avec absence de production protéique), des essais en phases précoces sont en cours. Il s’agit de traitements purement suspensifs à prendre à vie dont le coût est extrêmement élevé et dont il conviendra d’évaluer l’impact sur l’utilisation des autres traitements.16
La médecine personnalisée a transformé la prise en charge
L’identification du canal épithélial CFTR et la classification des 2 000 mutations environ du gène CFTR en diverses classes fonctionnelles ont permis d’introduire une médecine personnalisée en utilisant un potentiateur et divers correcteurs du canal CFTR. Les multiples essais randomisés ont prouvé leur efficacité sur la qualité de vie, la concentration chlorée sudorale, la fonction respiratoire, la diminution des exacerbations pulmonaires, les hospitalisations et surtout les indications de transplantation pulmonaire. Reste maintenant à démontrer leur efficacité sur la survie et leur tolérance, notamment hépatique, à long terme.
1. Registre français de la mucoviscidose. Bilan des données 2020. Vaincre la mucoviscidose. Paris, janvier 2022.
2. Rang C, Keating D, Wilson J, Kotsimbos T. Re-imagining cystic fibrosis care: next generation thinking. Eur Respir J 2020;55(5):1902443.
3. Elborn JS. Cystic fibrosis. Lancet 2016;388(10059):2519-31.
4. Nkam L, Lambert J, Latouche A, Bellis G, Burgel PR, Hocine MN. A 3-year prognostic score for adults with cystic fibrosis. J Cyst Fibros 2017;16(6):702-8.
5. Coriati A, Sykes J, Nkam L, Hocine MN, Burgel PR, Stephenson AL. Validation of the French 3-year prognostic score using the Canadian Cystic Fibrosis registry. J Cyst Fibros 2019;18(3):396-8.
6. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365(18):1663-72.
7. Davies JC, Wainwright CE, Canny GJ, Chilvers JA, Howenstine MS, Munck A, et al., on behalf of the VX08-770-103 (ENVISION) Study Group. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Resp Crit Care Med 2013;187(11):1219-25.
8. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. for the TRAFFIC and TRANSPORT Study Groups. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015;373(3):220-31.
9. Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med 2017;377(21):2013-23.
10. Rowe SM, Daines C, Rigshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med 2017;377(21):2024-35.
11. Middleton PG, Mall MA, Drevineck P, Lands LC, McKone EF, Polineni D, et al. for the VX17-445-102 Group. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508delallele. N Engl J Med 2019;381(19):1809-19.
12. Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al., on behalf of the VX-17-445-103 Trial Group. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind randomised, phase 3 trial. Lancet 2019;394:1940-8.
13. Barry PJ, Mall MA, Alvarez A, Colombo C, de Winter-de Groote KM, Fajac I, et al. for the VX18-445-104 Study Group. Triple therapy for cystic fibrosis Phe508del-gating and -residual function genotypes. N Engl J Med 2021;385(9):815-25.
14. Sutharsan S, McKone EF, Downey DG, Duckers J, MacGregor G, Tullis E, et al. for the VX18-445-109 study group. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: a 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir Med 2022;10(3):267-77.
15. Martin C, Burnet E, Ronayette-Preira A, de Carli P, Martin J, Delmas L, et al. Patient perspectives following initiation of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis and advanced lung disease. Respir Med Res 2021;80:100829.
16. Durieu I, Dalon F, Reynaud Q, Lemonnier L, Dehillotte C, Berard M, et al. Temporal trends in healthcare resource use and associated costs of patients with cystic fibrosis. J Cyst Fibros 2022;21(1):88-95.