La cholangite biliaire primitive, anciennement appelée cirrhose biliaire primitive, est une maladie cholestatique chronique d’origine auto-immune présumée, caractérisée par une atteinte inflammatoire des petits canaux biliaires, conduisant à leur destruction progressive (paucité biliaire). En l’absence de traitement, une fibrose hépatique extensive se constitue, aboutissant à la cirrhose et ses complications.
La cholangite biliaire primitive (CBP) est la première cause de cholestase chronique chez l’adulte mais reste néanmoins une maladie rare. Sa prévalence est estimée entre 10 et 40 pour 100 000 habitants (environ 20/100 000 en France) et son incidence annuelle entre 1 et 2 pour 100 000.1 Cette maladie atteint très préférentiellement les femmes (90 % des cas), d’un âge médian de 50 ans au moment du diagnostic. Elle est fréquemment associée à d’autres maladies auto-immunes, comme le syndrome de Gougerot-Sjögren (7 -34 % des cas), la thyroïdite de Hashimoto (11-13 %) ou le syndrome de Raynaud (9-13 %).2
La physiopathologie de cette maladie rare est encore mal connue
La physiopathologie de la CBP est complexe.3 Elle met en jeu l’immunité innée et adaptative dirigée spécifiquement contre des antigènes de la membrane mitochondriale, exprimés anormalement à la surface des cellules biliaires (cholangiocytes), avec intervention d’auto-anticorps (anticorps anti-mitochondrie [AAM]) mais également de lymphocytes T cytotoxiques. Il est actuellement admis que la diminution de la sécrétion cholangiocytaire de bicarbonates (qui assure une protection vis-à-vis de la toxicité cellulaire intrinsèque des acides biliaires) joue un rôle précoce majeur, en facilitant l’entrée en sénescence et en apoptose des cholangiocytes et l’expression aberrante d’antigènes membranaires. La réaction immunitaire entraîne une inflammation cholangiocytaire chronique, qui aboutit à une destruction progressive des petits canaux biliaires et à une accumulation intra-hépatique des acides biliaires, définissant la cholestase. Cette accumulation des acides biliaires est toxique pour les hépatocytes, et est à l’origine de la fibrose hépatique.
Comme dans toutes les maladies dites « auto-immunes », des facteurs génétiques et environnementaux semblent intervenir. Plusieurs gènes de susceptibilité ont été identifiés, incluant les gènes du complexe majeur d’histocompatibilité (HLA) ou des gènes de régulation de la production des cytokines pro-inflammatoires (IL-12). Il existe en outre des cas familiaux de cholangite biliaire primitive. Des facteurs infectieux ont aussi été suspectés : infections urinaires à répétition [mimétisme moléculaire entre les sous-unités CPD-E2 d’Escherichia coli et de la mitochondrie humaine (pathogen-associated molecular patterns – PAMPs)] voire une infection chronique par un β-rétrovirus. D’autres facteurs environnementaux ont été évoqués, notamment le tabagisme et le fait de vivre près d’usines de traitement de déchets. Ces hypothèses étiologiques ne sont pas exclusives et, globalement, la physiopathologie précise de la CBP comporte toujours beaucoup d’inconnues.
Comme dans toutes les maladies dites « auto-immunes », des facteurs génétiques et environnementaux semblent intervenir. Plusieurs gènes de susceptibilité ont été identifiés, incluant les gènes du complexe majeur d’histocompatibilité (HLA) ou des gènes de régulation de la production des cytokines pro-inflammatoires (IL-12). Il existe en outre des cas familiaux de cholangite biliaire primitive. Des facteurs infectieux ont aussi été suspectés : infections urinaires à répétition [mimétisme moléculaire entre les sous-unités CPD-E2 d’Escherichia coli et de la mitochondrie humaine (pathogen-associated molecular patterns – PAMPs)] voire une infection chronique par un β-rétrovirus. D’autres facteurs environnementaux ont été évoqués, notamment le tabagisme et le fait de vivre près d’usines de traitement de déchets. Ces hypothèses étiologiques ne sont pas exclusives et, globalement, la physiopathologie précise de la CBP comporte toujours beaucoup d’inconnues.
Quand évoquer et comment poser le diagnostic de CBP ?
Les signes cliniques les plus fréquents lors du diagnostic sont le prurit (20-50 % des cas) et l’asthénie (40-80 % des cas). Cependant, la maladie est souvent initialement asymptomatique et l’absence de signe clinique ne doit pas faire éliminer le diagnostic. Très rarement, la CBP est révélée par une complication de la cirrhose et sa présentation clinique est alors dominée par les signes d’insuffisance hépatocellulaire ou d’hypertension portale (ascite, hémorragie digestive par rupture de varices œsophagiennes, ictère).
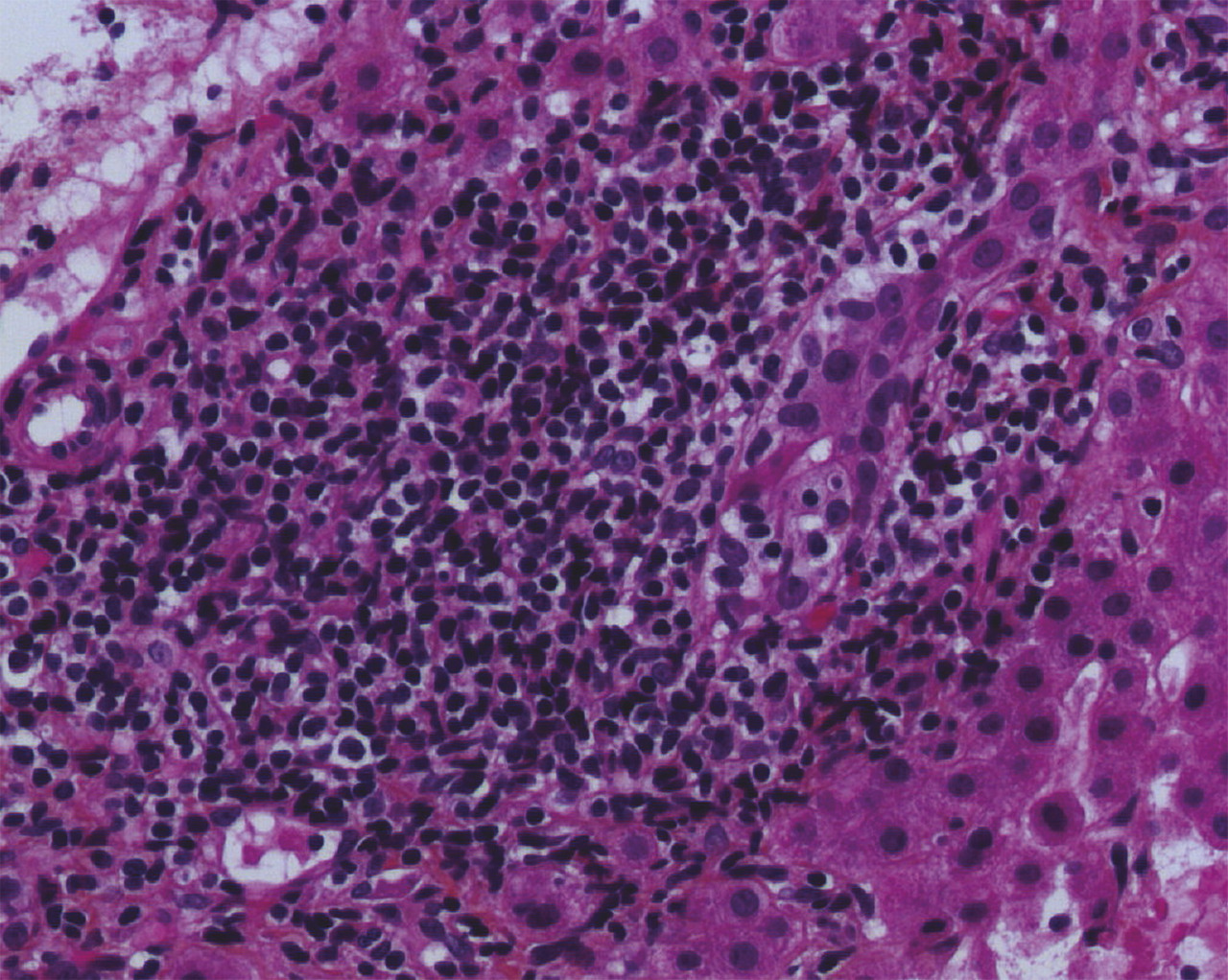
Biologiquement, il existe une cholestase chronique, définie par une augmentation concomitante des phosphatases alcalines (PAL) et de la gammaglutamyl transpeptidase (GGT) depuis au moins 6 mois. La cholestase peut être isolée, ou associée à une élévation modérée des transaminases et, plus rarement, à un ictère, qui est un signe de sévérité. Une augmentation des immunoglobulines sériques de type M (IgM) est fréquente. Le marqueur le plus spécifique de la CBP est la présence d’anticorps anti-mitochondrie de type M2 (AAM-2), à un taux sérique supérieur ou égal à 1/40e, présents chez plus de 95 % des patients atteints de CBP. Quand la biopsie hépatique est réalisée, on peut parfois observer des lésions de cholangite destructrice lymphocytaire ou granulomateuse, associées à un infiltrat portal et périportal lympho-plasmocytaire, éventuellement organisé en granulome, et parfois à une ductopénie (perte des canaux biliaires dans plus de 50 % des espaces portes) et à une fibrose (fig. 1 ).
Une forme particulière de cholangite biliaire primitive est l’association à une hépatite auto-immune (HAI) dans 5 à 15 % des cas, constituant un syndrome de chevauchement (ou overlap syndrome). La présentation clinique est identique à celle d’une CBP classique, mais l’élévation des transaminases (> 5N) et des IgG (> 20 g/L) est plus marquée et des anticorps anti-muscle lisse sont souvent présents. La biopsie hépatique est indispensable au diagnostic d’overlap et montre des lésions d’hépatite d’interface lympho-plasmocytaire (activité nécrotico-inflammatoire péri-portale) marquées, avec les lésions biliaires de la CBP.
Biologiquement, il existe une cholestase chronique, définie par une augmentation concomitante des phosphatases alcalines (PAL) et de la gammaglutamyl transpeptidase (GGT) depuis au moins 6 mois. La cholestase peut être isolée, ou associée à une élévation modérée des transaminases et, plus rarement, à un ictère, qui est un signe de sévérité. Une augmentation des immunoglobulines sériques de type M (IgM) est fréquente. Le marqueur le plus spécifique de la CBP est la présence d’anticorps anti-mitochondrie de type M2 (AAM-2), à un taux sérique supérieur ou égal à 1/40e, présents chez plus de 95 % des patients atteints de CBP. Quand la biopsie hépatique est réalisée, on peut parfois observer des lésions de cholangite destructrice lymphocytaire ou granulomateuse, associées à un infiltrat portal et périportal lympho-plasmocytaire, éventuellement organisé en granulome, et parfois à une ductopénie (perte des canaux biliaires dans plus de 50 % des espaces portes) et à une fibrose (
Une forme particulière de cholangite biliaire primitive est l’association à une hépatite auto-immune (HAI) dans 5 à 15 % des cas, constituant un syndrome de chevauchement (ou overlap syndrome). La présentation clinique est identique à celle d’une CBP classique, mais l’élévation des transaminases (> 5N) et des IgG (> 20 g/L) est plus marquée et des anticorps anti-muscle lisse sont souvent présents. La biopsie hépatique est indispensable au diagnostic d’overlap et montre des lésions d’hépatite d’interface lympho-plasmocytaire (activité nécrotico-inflammatoire péri-portale) marquées, avec les lésions biliaires de la CBP.
Critères diagnostiques de CBP
Le diagnostic positif de CBP repose sur l’association de 2 des 3 critères suivants (tableau 1 )4 :
– signes biologiques de cholestase chronique, définis par une augmentation simultanée des PAL et de la GGT depuis au moins 6 mois ;
– détection d’auto-anticorps spécifiques de la maladie : anticorps anti-mitochondrie ≥ 1/40 en immunofluorescence indirecte (IFI), confirmés en immunoblot ou Elisa (sensibilité > 90 %, spécificité > 95 %), et/ou anticorps antinucléaires spécifiques, de type cerclés (correspondant le plus souvent à des anticorps anti-gp210) ou de type « multiple nuclear dots » (anticorps anti-Sp100, sensibilité ≤ 25 %, spécificité > 90 %) ;
– lésions de cholangite destructrice, granulomateuse ou lymphocytaire, sur l’histologie hépatique (observées dans seulement 30 % à 50 % des biopsies percutanées du fait de l’hétérogénéité lésionnelle).
Dans la majorité des cas, l’examen histologique du foie n’est pas nécessaire au diagnostic de la cholangite biliaire primitive. La biopsie hépatique doit être proposée à titre diagnostique en l’absence d’auto-anticorps spécifiques de CBP, ou si l’on suspecte un syndrome de chevauchement ou toute autre comorbidité hépatique (syndrome métabolique, surpoids, alcool, etc.).
Il est important de noter que la présence isolée (c’est-à-dire sans anomalie associée des tests hépatiques) d’auto-anticorps spécifiques de la CBP ne permet pas de porter le diagnostic de cholangite biliaire primitive. Dans cette situation, une surveillance annuelle des tests hépatiques est recommandée : seuls 15 % des patients concernés développeront à 5 ans une élévation des PAL, permettant alors de confirmer le diagnostic.
Le bilan initial doit avoir écarté, par échographie, toute cause de cholestase extra-hépatique. La cholangio-IRM (normale dans la CBP) n’est pas nécessaire, sauf dans les formes atypiques (absence d’auto-anticorps caractéristiques) ou en cas de doute diagnostique avec une cholangite sclérosante primitive.
– signes biologiques de cholestase chronique, définis par une augmentation simultanée des PAL et de la GGT depuis au moins 6 mois ;
– détection d’auto-anticorps spécifiques de la maladie : anticorps anti-mitochondrie ≥ 1/40 en immunofluorescence indirecte (IFI), confirmés en immunoblot ou Elisa (sensibilité > 90 %, spécificité > 95 %), et/ou anticorps antinucléaires spécifiques, de type cerclés (correspondant le plus souvent à des anticorps anti-gp210) ou de type « multiple nuclear dots » (anticorps anti-Sp100, sensibilité ≤ 25 %, spécificité > 90 %) ;
– lésions de cholangite destructrice, granulomateuse ou lymphocytaire, sur l’histologie hépatique (observées dans seulement 30 % à 50 % des biopsies percutanées du fait de l’hétérogénéité lésionnelle).
Dans la majorité des cas, l’examen histologique du foie n’est pas nécessaire au diagnostic de la cholangite biliaire primitive. La biopsie hépatique doit être proposée à titre diagnostique en l’absence d’auto-anticorps spécifiques de CBP, ou si l’on suspecte un syndrome de chevauchement ou toute autre comorbidité hépatique (syndrome métabolique, surpoids, alcool, etc.).
Il est important de noter que la présence isolée (c’est-à-dire sans anomalie associée des tests hépatiques) d’auto-anticorps spécifiques de la CBP ne permet pas de porter le diagnostic de cholangite biliaire primitive. Dans cette situation, une surveillance annuelle des tests hépatiques est recommandée : seuls 15 % des patients concernés développeront à 5 ans une élévation des PAL, permettant alors de confirmer le diagnostic.
Le bilan initial doit avoir écarté, par échographie, toute cause de cholestase extra-hépatique. La cholangio-IRM (normale dans la CBP) n’est pas nécessaire, sauf dans les formes atypiques (absence d’auto-anticorps caractéristiques) ou en cas de doute diagnostique avec une cholangite sclérosante primitive.
Évaluer la sévérité : rôle central de l’élastométrie
La sévérité de la CBP (maladie avancée ou non) est évaluée au moment du diagnostic à l’aide de critères cliniques, biologiques et radiologiques, et par le degré de fibrose hépatique.
Les signes cliniques et biologiques de cirrhose doivent être recherchés : signes d’insuffisance hépatocellulaire (angiomes stellaires, ictère, encéphalopathie hépatique, albuminémie < 35 g/L, TP < 50 %) et d’hypertension portale (circulation veineuse collatérale, ascite, numération plaquettaire < 150 000/mm3).
Au moment du diagnostic :
– toute élévation de la bilirubine totale et conjuguée ou des PAL > 5N est le signe d’une maladie avancée et habituellement symptomatique ;
– un rapport ASAT/plaquettes (score APRI) > 0,54 est associé à un risque plus élevé de complications ;
– les anticorps anti-gp210 et anti-sp100 sont associés à des formes plus avancées de CBP.
L’écho-Doppler hépatique recherche des signes de cirrhose : dysmorphie hépatique, signes d’hypertension portale directs (élargissement du tronc porte, diminution du flux portal en Doppler, voies de dérivation porto-systémiques) ou indirects (splénomégalie, ascite). L’échographie permet également de vérifier l’absence de nodule hépatique évocateur de carcinome hépatocellulaire.
L’élastométrie hépatique (FibroScan) permet d’évaluer le degré de fibrose hépatique en mesurant la dureté du foie. Elle est recommandée pour l’évaluation initiale de la gravité de la CBP et doit remplacer la biopsie hépatique chaque fois que cela est possible. Une valeur d’élastométrie > 10,7 kPa est corrélée à une fibrose sévère et > 16,9 kPa à la cirrhose. Cet examen non invasif occupe désormais une place majeure dans l’évaluation de la sévérité de la CBP.
La biopsie du foie n’est recommandée, pour évaluer la fibrose hépatique, qu’en cas d’échec ou de discordance répétée de l’élastométrie. Plusieurs classifications histologiques peuvent être utilisées pour estimer le stade de la maladie.
Le dépistage des varices œsophagiennes et gastriques par endoscopie œso-gastro-duodénale est indiqué en cas de numération plaquettaire < 150 000/mm3 ou d’élastométrie > 20 KPa (critères de Baveno VI).
Les signes cliniques et biologiques de cirrhose doivent être recherchés : signes d’insuffisance hépatocellulaire (angiomes stellaires, ictère, encéphalopathie hépatique, albuminémie < 35 g/L, TP < 50 %) et d’hypertension portale (circulation veineuse collatérale, ascite, numération plaquettaire < 150 000/mm3).
Au moment du diagnostic :
– toute élévation de la bilirubine totale et conjuguée ou des PAL > 5N est le signe d’une maladie avancée et habituellement symptomatique ;
– un rapport ASAT/plaquettes (score APRI) > 0,54 est associé à un risque plus élevé de complications ;
– les anticorps anti-gp210 et anti-sp100 sont associés à des formes plus avancées de CBP.
L’écho-Doppler hépatique recherche des signes de cirrhose : dysmorphie hépatique, signes d’hypertension portale directs (élargissement du tronc porte, diminution du flux portal en Doppler, voies de dérivation porto-systémiques) ou indirects (splénomégalie, ascite). L’échographie permet également de vérifier l’absence de nodule hépatique évocateur de carcinome hépatocellulaire.
L’élastométrie hépatique (FibroScan) permet d’évaluer le degré de fibrose hépatique en mesurant la dureté du foie. Elle est recommandée pour l’évaluation initiale de la gravité de la CBP et doit remplacer la biopsie hépatique chaque fois que cela est possible. Une valeur d’élastométrie > 10,7 kPa est corrélée à une fibrose sévère et > 16,9 kPa à la cirrhose. Cet examen non invasif occupe désormais une place majeure dans l’évaluation de la sévérité de la CBP.
La biopsie du foie n’est recommandée, pour évaluer la fibrose hépatique, qu’en cas d’échec ou de discordance répétée de l’élastométrie. Plusieurs classifications histologiques peuvent être utilisées pour estimer le stade de la maladie.
Le dépistage des varices œsophagiennes et gastriques par endoscopie œso-gastro-duodénale est indiqué en cas de numération plaquettaire < 150 000/mm3 ou d’élastométrie > 20 KPa (critères de Baveno VI).
Dépister les maladies associées, notamment auto-immunes
Une autre maladie auto-immune est présente chez 50 % des patients atteints de CBP.5 Il est donc important de dépister, lors du diagnostic de la cholangite biliaire primitive, d’éventuelles pathologies auto-immunes associées : thyroïdite de Hashimoto, syndrome de Gougerot-Sjögren, syndrome de Raynaud, sclérodermie, maladie cœliaque… Le bilan initial devra donc comporter, au minimum, un interrogatoire, un examen clinique à la recherche de manifestations auto-immunes (syndrome sec, douleurs articulaires, signes cutanés), et un dosage de la TSH. Le syndrome de chevauchement avec l’hépatite auto-immune (forme mixte) fait partie, quant à lui, du bilan diagnostique de la maladie.
Par ailleurs, la CBP est associée à un risque d’ostéoporose fracturaire multiplié par 3, imposant la réalisation d’une ostéodensitométrie dès le diagnostic, à renouveler au cours du suivi tous les 2 à 4 ans.5 Enfin, une hypercholestérolémie est fréquente, portant souvent plus sur les HDL que sur les LDL, son traitement devra s’intégrer dans la prise en charge du risque cardiovasculaire global.
Par ailleurs, la CBP est associée à un risque d’ostéoporose fracturaire multiplié par 3, imposant la réalisation d’une ostéodensitométrie dès le diagnostic, à renouveler au cours du suivi tous les 2 à 4 ans.5 Enfin, une hypercholestérolémie est fréquente, portant souvent plus sur les HDL que sur les LDL, son traitement devra s’intégrer dans la prise en charge du risque cardiovasculaire global.
Le pronostic s’est considérablement amélioré
La CBP évolue lentement, sur plusieurs années voire dizaines d’années. La destruction progressive des petits canaux biliaires entraîne le développement de la fibrose puis de la cirrhose et de ses complications. Les premières séries descriptives de patients atteints de CBP, étudiées avant l’arrivée des traitements efficaces, montraient un pronostic défavorable avec une progression rapide de la fibrose et une évolution vers la cirrhose en moins de 4 ans, pour une survie médiane de 6 à 10 ans.6 L’introduction de l’acide ursodésoxycholique (AUDC), dans les années 1990, a considérablement amélioré ce pronostic.
Plusieurs critères cliniques, souvent associés à une moins bonne réponse biologique à l’AUDC, ont pu être reliés à un moins bon pronostic à long terme : l’âge jeune au diagnostic (< 45 ans), le sexe masculin, la présence d’un prurit sévère ou d’un ictère. Un stade histologique avancé (fibrose hépatique extensive ou cirrhose) ou une élastométrie hépatique > 9,6 kPa constituent également des facteurs pronostiques péjoratifs. La présence de signes évocateurs d’hépatite auto-immune (syndrome de chevauchement) pourrait également être associée à un moins bon pronostic. Indépendamment de tous ces facteurs, la réponse biologique à l’AUDC, évaluée après 6 à 12 mois de traitement, est aujourd’hui reconnue comme le principal marqueur pronostique de la CBP. Plusieurs définitions de la réponse biologique à l’AUDC ont été proposées pour aider le clinicien à identifier les patients à risque (tableau 2 ). Les critères les plus utilisés en pratique clinique sont ceux de Toronto et de Paris-II.
Plusieurs critères cliniques, souvent associés à une moins bonne réponse biologique à l’AUDC, ont pu être reliés à un moins bon pronostic à long terme : l’âge jeune au diagnostic (< 45 ans), le sexe masculin, la présence d’un prurit sévère ou d’un ictère. Un stade histologique avancé (fibrose hépatique extensive ou cirrhose) ou une élastométrie hépatique > 9,6 kPa constituent également des facteurs pronostiques péjoratifs. La présence de signes évocateurs d’hépatite auto-immune (syndrome de chevauchement) pourrait également être associée à un moins bon pronostic. Indépendamment de tous ces facteurs, la réponse biologique à l’AUDC, évaluée après 6 à 12 mois de traitement, est aujourd’hui reconnue comme le principal marqueur pronostique de la CBP. Plusieurs définitions de la réponse biologique à l’AUDC ont été proposées pour aider le clinicien à identifier les patients à risque (
Plusieurs lignes de traitement
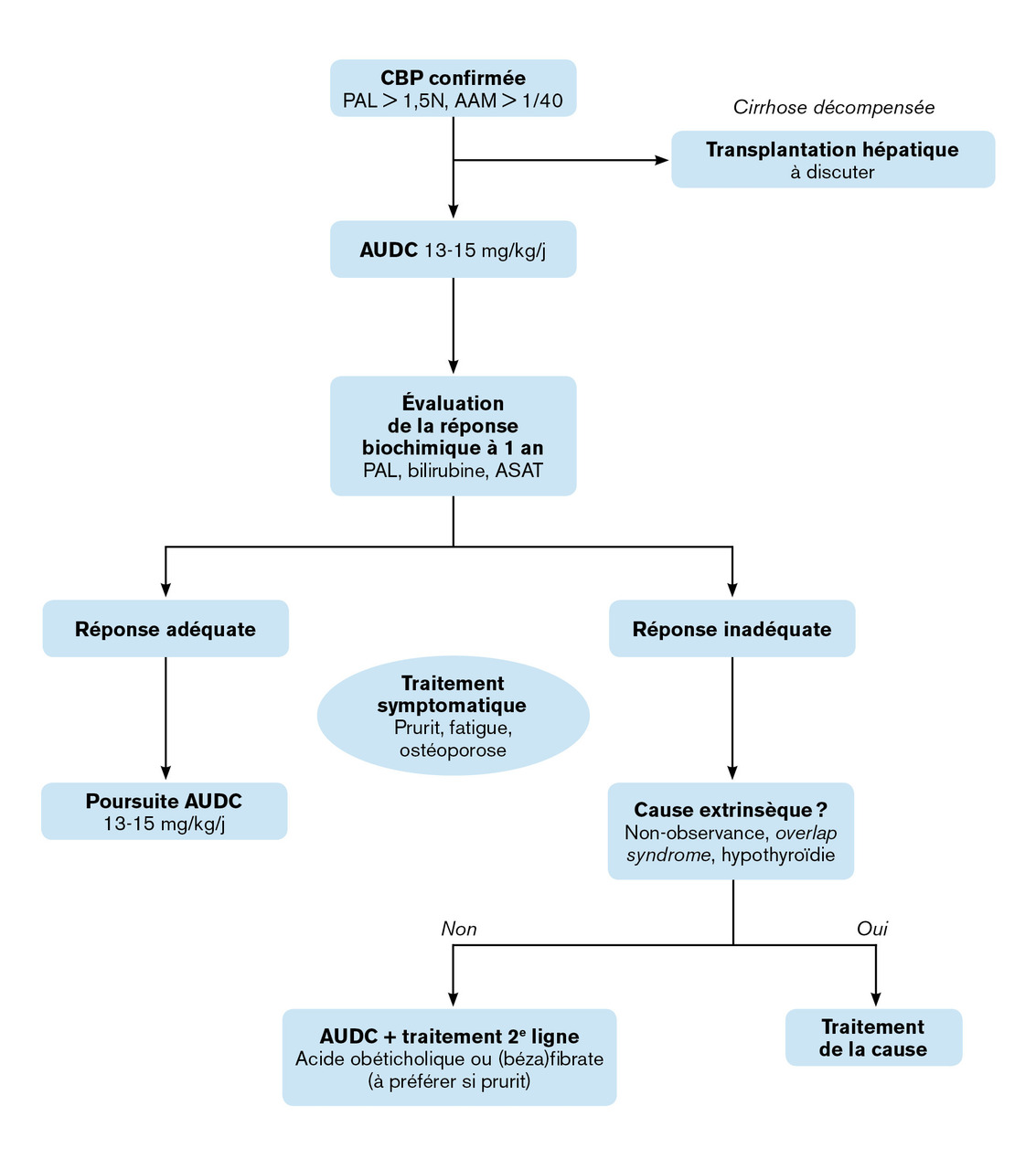
Des traitements efficaces sont aujourd’hui disponibles, dont les indications sont bien définies (fig. 2 )
L’acide ursodésoxycholique efficace dans 60 à 70 % des cas
L’AUDC est le premier traitement de la cholangite biliaire primitive dont l’efficacité a été démontrée dans les années 1990.7 Il s’agit d’un acide biliaire hydrophile non cytotoxique ayant des propriétés à la fois cholérétiques, anti-inflammatoires et immunomodulatrices. Administré quotidiennement par voie orale, il permet de réduire la quantité des acides biliaires endogènes circulants, protège le foie de l’action cytotoxique de ces acides biliaires, et diminue la réaction inflammatoire associée à la cholestase. L’AUDC a montré son efficacité pour améliorer la survie sans transplantation (78-80 % à 10 ans sous AUDC contre 59-60 % en l’absence de traitement), pour réduire la progression de la fibrose hépatique et de l’hypertension portale, et pour améliorer, de façon quasi constante, les tests biologiques hépatiques. L’AUDC, à la dose de 13 à 15 mg/kg/j, est recommandé comme traitement de première intention de la CBP.4 Il est généralement bien toléré, mais une diarrhée et une majoration transitoire du prurit peuvent apparaître en début de traitement.
La réponse à l’acide ursodésoxycholique doit être évaluée classiquement à un an, sur des critères biologiques devant inclure au minimum la bilirubine et les PAL (tableau 2 ). Sur la base des critères les plus communément utilisés (critères de Paris-II, critères de Toronto), environ 30 à 40 % des patients ont une réponse à l’AUDC considérée comme insuffisante, c’est-à-dire associée à un risque significativement plus élevé de complications, de décès ou de transplantation hépatique.8
La réponse à l’acide ursodésoxycholique doit être évaluée classiquement à un an, sur des critères biologiques devant inclure au minimum la bilirubine et les PAL (
Traitements anti-cholestatiques de 2e intention
Plusieurs traitements complémentaires sont actuellement disponibles pour traiter les patients dont la réponse à l’acide ursodésoxycholique est insuffisante. L’acide obéticholique est un agoniste du récepteur nucléaire des acides biliaires Farnesoid-X receptor (FXR), régulateur majeur de l’homéostasie biliaire. Il diminue la synthèse hépatique des acides biliaires et facilite leur excrétion biliaire. Son efficacité a été montrée en termes de réduction des anomalies des tests hépatiques, à un an, par un essai publié en 2016.9 Son principal effet secondaire est le prurit ; rapporté par les deux tiers des patients, il peut parfois conduire à interrompre le traitement, malgré une adaptation des doses. L’acide obéticholique a obtenu une autorisation de mise sur le marché (AMM) conditionnelle pour traiter les patients intolérants ou dont la réponse à l’AUDC est insuffisante. Son bénéfice sur la survie reste à démontrer.
Les fibrates (agonistes des récepteurs PPARα), classiquement utilisés comme hypolipémiants, ont aussi prouvé leur efficacité dans le traitement de 2e ligne de la cholangite biliaire primitive. Ils jouent un rôle dans la diminution de la synthèse des acides biliaires et dans la production de facteurs anti-inflammatoires au niveau hépatique. L’efficacité du bézafibrate a été évaluée par un essai randomisé publié en 2018, montrant une réduction des anomalies des tests hépatiques, à 2 ans, avec une normalisation des PAL chez 67 % des personnes traitées.10 Cet effet biologique est associé à une diminution du prurit et à une moindre progression des marqueurs de fibrose, dont l’élastométrie. Les principaux effets secondaires du bézafibrate sont les myalgies (20 % des cas dans l’essai BEZURSO), qui imposent parfois l’arrêt du traitement. Une élévation de moins de 5 % du taux de créatinine est observée sous fibrates, mais sans insuffisance rénale et sans qu’il soit nécessaire d’arrêter le traitement. Les fibrates n’ont pas encore d’AMM dans le traitement de la CBP. Comme pour l’acide obéticholique, leur efficacité sur la survie, pour le moment, n’est pas établie.
Aucune étude actuellement disponible ne compare l’efficacité et la tolérance des fibrates à celles de l’acide obéticholique en 2e ligne de traitement de la CBP. Le choix du traitement doit tenir compte des symptômes (prurit, myalgies), des effets secondaires respectifs de chacune des molécules et de leurs coûts (élevé pour l’acide obéticholique, très faible pour les fibrates).
Les fibrates (agonistes des récepteurs PPARα), classiquement utilisés comme hypolipémiants, ont aussi prouvé leur efficacité dans le traitement de 2e ligne de la cholangite biliaire primitive. Ils jouent un rôle dans la diminution de la synthèse des acides biliaires et dans la production de facteurs anti-inflammatoires au niveau hépatique. L’efficacité du bézafibrate a été évaluée par un essai randomisé publié en 2018, montrant une réduction des anomalies des tests hépatiques, à 2 ans, avec une normalisation des PAL chez 67 % des personnes traitées.10 Cet effet biologique est associé à une diminution du prurit et à une moindre progression des marqueurs de fibrose, dont l’élastométrie. Les principaux effets secondaires du bézafibrate sont les myalgies (20 % des cas dans l’essai BEZURSO), qui imposent parfois l’arrêt du traitement. Une élévation de moins de 5 % du taux de créatinine est observée sous fibrates, mais sans insuffisance rénale et sans qu’il soit nécessaire d’arrêter le traitement. Les fibrates n’ont pas encore d’AMM dans le traitement de la CBP. Comme pour l’acide obéticholique, leur efficacité sur la survie, pour le moment, n’est pas établie.
Aucune étude actuellement disponible ne compare l’efficacité et la tolérance des fibrates à celles de l’acide obéticholique en 2e ligne de traitement de la CBP. Le choix du traitement doit tenir compte des symptômes (prurit, myalgies), des effets secondaires respectifs de chacune des molécules et de leurs coûts (élevé pour l’acide obéticholique, très faible pour les fibrates).
Place de la corticothérapie et des traitements immunosuppresseurs
En dehors des formes mixtes de CBP (syndrome de chevauchement), l’intérêt de la corticothérapie ou des immunosuppresseurs, comme l’azathioprine ou le mycophénolate mofétil, n’est pas démontrée. Toutefois, il existe un faisceau d’arguments en faveur de l’efficacité potentielle du budésonide, prescrit en association à l’AUDC, à la posologie orale de 6 mg par jour, chez certains patients ayant une réponse insuffisante à l’AUDC et/ou des lésions inflammatoires histologiques significativement marquées. Le budésonide, bénéficiant d’un effet de premier passage hépatique important, a peu d’effets secondaires systémiques comparé aux corticoïdes classiques. Plusieurs études préliminaires ont montré qu’il était capable d’améliorer les signes biologiques et histologiques de la maladie. Un essai plus récent, encore non publié, a montré un effet bénéfique sur les PAL mais n’a pas confirmé la régression des signes histologiques de la maladie. La prescription de budésonide en 2e (voire 3e) ligne de traitement doit être décidée dans un centre expert.
Associés à l’AUDC, les corticoïdes systémiques constituent en revanche le traitement de choix des formes mixtes associant CBP et hépatite auto-immune (overlap syndrome). Ce traitement combiné peut être proposé d’emblée, si les signes d’hépatite auto-immune sont francs, ou si la réponse biologique est insuffisante au terme d’une période test de 6 mois d’un traitement par AUDC seul. Si la poursuite à long terme (≥ 12 mois) des corticoïdes s’avère nécessaire (corticodépendance), un traitement immunosuppresseur d’épargne des stéroïdes, tel que l’azathioprine ou le mycophénolate mofétil, peut être considéré.
Associés à l’AUDC, les corticoïdes systémiques constituent en revanche le traitement de choix des formes mixtes associant CBP et hépatite auto-immune (overlap syndrome). Ce traitement combiné peut être proposé d’emblée, si les signes d’hépatite auto-immune sont francs, ou si la réponse biologique est insuffisante au terme d’une période test de 6 mois d’un traitement par AUDC seul. Si la poursuite à long terme (≥ 12 mois) des corticoïdes s’avère nécessaire (corticodépendance), un traitement immunosuppresseur d’épargne des stéroïdes, tel que l’azathioprine ou le mycophénolate mofétil, peut être considéré.
La greffe en dernier recours
La transplantation hépatique (TH) reste le traitement de dernier recours, en cas de cirrhose décompensée ou de carcinome hépatocellulaire. Un taux de bilirubine > 80 µmol/L pendant plus de 6 mois doit faire envisager une TH même en l’absence de toute autre complication. Un prurit sévère, réfractaire à tout traitement médical, reste une indication exceptionnelle. La CBP est désormais une indication relativement rare de transplantation hépatique. Elle ne constituait que 3 % des indications de greffe en France en 2018 (37 sur 1 325 greffes). En Europe, on compte environ 200 patients greffés pour CBP par an ; les résultats sont excellents avec une survie globale de 80 % à 5 ans. Néanmoins, la récidive de la CBP sur le greffon est fréquente ; elle concerne environ un tiers des patients à 10 ans, et est associée à une diminution de la survie, du greffon et des patients. Une étude rétrospective récente, incluant près de 800 patients suivis en moyenne 11 ans après une transplantation hépatique pour CBP, a montré que l’administration préventive d’AUDC après la greffe était associée à une diminution des risques de récidive de la maladie, de perte du greffon et de décès.
Traiter les symptômes
Outre la prise en charge spécifique de la maladie, le traitement des symptômes est essentiel pour maintenir une qualité de vie la plus optimale possible. Le prurit doit être géré selon une cascade thérapeutique en commençant par la cholestyramine (à prendre à distance, de plusieurs heures, de l’AUDC), puis le bézafibrate, la rifampicine, la naltrexone et la sertraline. En cas de forme sévère résistante à ces traitements, il peut nécessiter le recours à des techniques d’épuration sanguine extracorporelle (échanges plasmatiques, système MARS) ou d’interruption transitoire du cycle entéro-hépatique (drainage naso-biliaire). La fatigue chronique (symptôme très fréquent) doit faire rechercher des causes potentiellement curables (dysthyroïdie, maladie cœliaque, anémie, diabète, dépression). Le syndrome sec peut justifier une prise en charge spécifique (larmes et salives artificielles, sialagogue). Enfin, l’ostéoporose et le risque fracturaire doivent être évalués et pris en charge.
Le rythme de la surveillance dépend de différents paramètres
Le rythme de surveillance doit être adapté à la sévérité initiale de la CBP, et à la réponse aux différents traitements (tous les 6 à 12 mois en cas de forme peu sévère, tous les 3 mois en cas de forme symptomatique ou sévère). Cette surveillance doit comprendre, au minimum, un examen clinique à la recherche d’un prurit ou d’un ictère, et au plan biologique, NFS, bilirubine, PAL, GGT, transaminases, albumine et TP. L’évolution de la fibrose hépatique doit être contrôlée par élastométrie tous les 12 mois si la réponse biologique est insuffisante, et tous les 2 à 3 ans en cas de bonne réponse. Le dosage de la TSH doit être renouvelé une fois par an, et l’ostéodensitométrie tous les 2 à 4 ans. En cas de cirrhose, le dépistage du carcinome hépatocellulaire par échographie doit être réalisé tous les 6 mois. Des varices œsophagiennes doivent être recherchées en cas de numération des plaquettes < 150 000/mm3 ou de chiffres d’élastométrie > 20 kPa (critères de Baveno VI).
Une prise en charge codifiée
La CBP, maladie cholestatique intrahépatique la plus fréquente chez l’adulte, atteint principalement les femmes de plus de 50 ans. Son diagnostic repose le plus souvent sur des critères biologiques et immunologiques, mais aussi parfois histologiques. L’administration au long cours d’acide ursodésoxycholique constitue le traitement de référence. En cas de réponse biologique insuffisante, un traitement de 2e ligne incluant soit l’acide obéticholique, soit le bézafibrate, doit lui être associé. Dans les formes les plus sévères, la transplantation hépatique est un traitement efficace.
Références
1. Dahlqvist G, Gaouar F, Carrat F, Meurisse S, Chazouillères O, Poupon R, et al. Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology. 2017;65(1):152‑63.
2. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. The Lancet. 2015;386(10003):1565‑75.
3. Gulamhusein AF, Hirschfield GM. Pathophysiology of primary biliary cholangitis. Best Practice & Research Clinical Gastroenterology. 2018;34‑35:17‑25.
4. Hirschfield GM, Beuers U, Corpechot C, Invernizzi P, Jones D, Marzioni M, et al. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. Journal of Hepatology. 2017;67(1):145‑72.
5. Corpechot C, Chrétien Y, Chazouillères O, Poupon R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. Journal of Hepatology. 2010;53(1):162‑9.
6. Prince M, Chetwynd A, Newman W, Metcalf JV, James OFW. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: Follow-up for up to 28 years. Gastroenterology. 2002;123(4):1044‑51.
7. Poupon RE, Balkau B, Eschwège E, Poupon R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Engl J Med. 1991;324(22):1548‑54.
8. Corpechot C, Chazouillères O, Poupon R. Early primary biliary cirrhosis: Biochemical response to treatment and prediction of long-term outcome. Journal of Hepatology. 2011;55(6):1361‑7.
9. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo- controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631‑43.
10. Corpechot C, Chazouillères O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A Placebo-controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N Engl J Med. 2018;378(23):2171‑81.
2. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. The Lancet. 2015;386(10003):1565‑75.
3. Gulamhusein AF, Hirschfield GM. Pathophysiology of primary biliary cholangitis. Best Practice & Research Clinical Gastroenterology. 2018;34‑35:17‑25.
4. Hirschfield GM, Beuers U, Corpechot C, Invernizzi P, Jones D, Marzioni M, et al. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. Journal of Hepatology. 2017;67(1):145‑72.
5. Corpechot C, Chrétien Y, Chazouillères O, Poupon R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. Journal of Hepatology. 2010;53(1):162‑9.
6. Prince M, Chetwynd A, Newman W, Metcalf JV, James OFW. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: Follow-up for up to 28 years. Gastroenterology. 2002;123(4):1044‑51.
7. Poupon RE, Balkau B, Eschwège E, Poupon R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Engl J Med. 1991;324(22):1548‑54.
8. Corpechot C, Chazouillères O, Poupon R. Early primary biliary cirrhosis: Biochemical response to treatment and prediction of long-term outcome. Journal of Hepatology. 2011;55(6):1361‑7.
9. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo- controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631‑43.
10. Corpechot C, Chazouillères O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A Placebo-controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N Engl J Med. 2018;378(23):2171‑81.
Dans cet article
- La physiopathologie de cette maladie rare est encore mal connue
- Quand évoquer et comment poser le diagnostic de CBP ?
- Dépister les maladies associées, notamment auto-immunes
- Le pronostic s’est considérablement amélioré
- Plusieurs lignes de traitement
- Le rythme de la surveillance dépend de différents paramètres
- Une prise en charge codifiée

Résumé
La cholangite biliaire primitive (ou anciennement cirrhose biliaire primitive, CBP) est la maladie cholestatique chronique la plus fréquente. Il s’agit d’une affection présumée auto-immune caractérisée par une inflammation et une destruction progressive des petits canaux biliaires, aboutissant, en l’absence de traitement, à une fibrose hépatique puis à une cirrhose. Elle atteint majoritairement les femmes (90 % des cas) de plus de 40 ans. Les signes cliniques révélateurs (prurit et asthénie) sont inconstants et peu spécifiques. Le diagnostic repose sur l’association d’une cholestase biologique (élévation concomitante des PAL et de la GGT) chronique et la présence d’auto-anticorps spécifiques (en particulier anti-mitochondrie de type 2). Le traitement de référence est l’acide ursodésoxycholique (AUDC) qui a fortement amélioré le pronostic de la CBP. Dans 30 à 40 % des cas, la réponse biologique à l’AUDC est insuffisante et des traitements complémentaires sont nécessaires, parmi lesquels figurent actuellement l’acide obéticholique et les fibrates.