L’ictère est un diagnostic clinique défini par une coloration jaune des téguments. Il est lié à l’augmentation anormale du taux de bilirubine plasmatique (> 40 µmol/L).
Ce pigment provient de la dégradation de l’hémoglobine par les macrophages. Son taux normal est de l’ordre de 20 µmol/L, associant bilirubine non conjuguée (< 15 µmol/L) et bilirubine conjuguée (< 5 µmol/L). Dans le plasma, la bilirubine totale est majoritairement non conjuguée, peu soluble et liée à l’albumine. Elle ne passe pas dans les urines. Elle est captée par l’hépatocyte via des transporteurs, l’albumine restant dans le plasma. La bilirubine est ensuite conjuguée à l’acide glucuronique dans l’hépatocyte grâce à la bilirubine glucuronyltransférase. Enfin, elle est excrétée dans la bile grâce à un transporteur situé au pôle canaliculaire de l’hépatocyte.
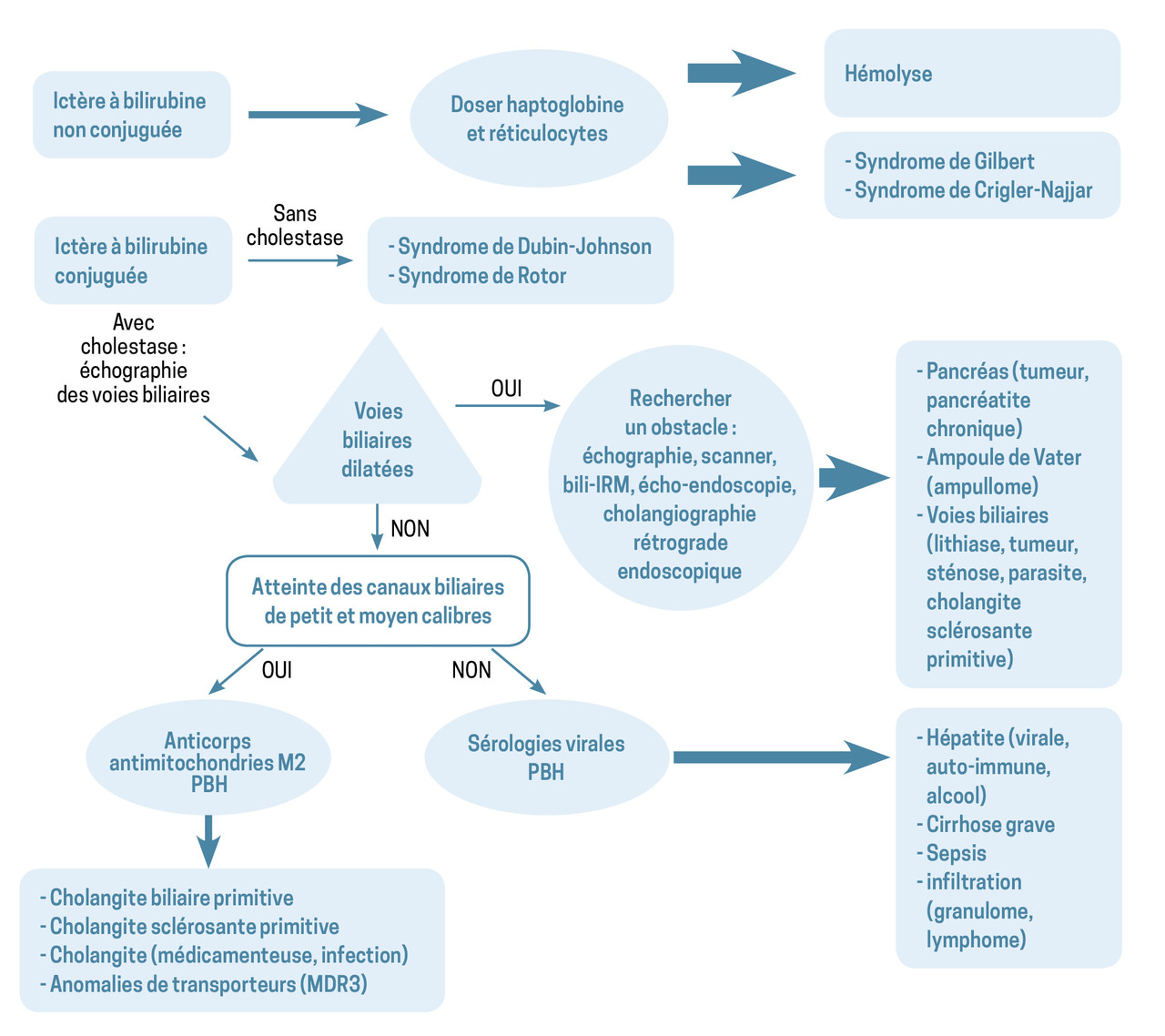
Ictère à bilirubine non conjuguée
Dans ces situations, les urines sont claires.
Différentes causes sont possibles. Les plus fréquentes à connaître sont au nombre de quatre.
Hémolyse et dysérythropoïèse
Un hémogramme et un dosage des réticulocytes et de l’haptoglobine sont nécessaires au diagnostic. À noter : en cas d’hémolyse1, l’anémie n’est pas systématique car elle peut être compensée.
Syndrome de Gilbert
Cette affection bénigne est liée à une anomalie du gène promoteur de la bilirubine glucuronyltransférase entraînant un déficit enzymatique partiel. Jusqu’à 10 % de la population a cette anomalie, transmise selon un mode autosomique récessif. Chez ces sujets, le jeûne et les infections favorisent l’élévation de la bilirubine (au maximum 80 µmol/L). Les autres tests hépatiques sont normaux.
Le phénobarbital ou le méprobamate, inducteurs enzymatiques, peuvent permettre de diminuer l’ictère.
Le syndrome de Gilbert étant bénin, le diagnostic génétique est exceptionnellement demandé.
Ictère néonatal (dit physiologique)
La maturation complète de l’activité de la bilirubine glucuronyltransférase est ici retardée.
L’ictère néonatal survient chez 65 à 70 % des nouveau-nés. Il disparaît spontanément en quelques jours.
Il faut cependant être attentif au risque d’ictère pathologique en éliminant les facteurs de risque suivants :
– survenue précoce (avant 24 heures de vie) ;
– signes d’hémolyse (splénomégalie, syndrome anémique) ;
– signe de choléstase (hépatomégalie, selles décolorées et urines foncées) ;
– durée supérieure à dix jours.
Maladie de Crigler-Najjar
Rare (incidence de 1/1 000 000 naissances), elle est liée au déficit complet de l’activité de la bilirubine glucuronyltransférase, du fait de mutations du gène de l’enzyme transmises selon un mode autosomique récessif. Dans ce cadre, l’ictère de survenue néonatale précoce et intense est potentiellement grave : risque d’encéphalopathie par toxicité cérébrale de la bilirubine non conjuguée.
Ictère à bilirubine conjuguée sans cholestase
Sont ici en cause des affections génétiques rares altérant l’excrétion de la bilirubine conjuguée depuis l’hépatocyte. Les deux principales sont le syndrome de Rotor (maladie héréditaire bénigne sans hémolyse ni anomalies histologiques hépatiques) et le syndrome de Dubin-Johnson (maladie héréditaire bénigne avec des anomalies histologiques à type de dépôt de pigments brun-noir dans les cellules parenchymateuses hépatiques).
Ictère à bilirubine conjuguée avec cholestase
En cas de cholestase, il existe une diminution du flux biliaire. La bilirubine conjuguée reflue alors dans le plasma ; hydrosoluble, non liée à l’albumine, elle passe dans les urines.
Quand y penser ?
Les urines sont foncées. Le patient peut se plaindre d’un prurit.
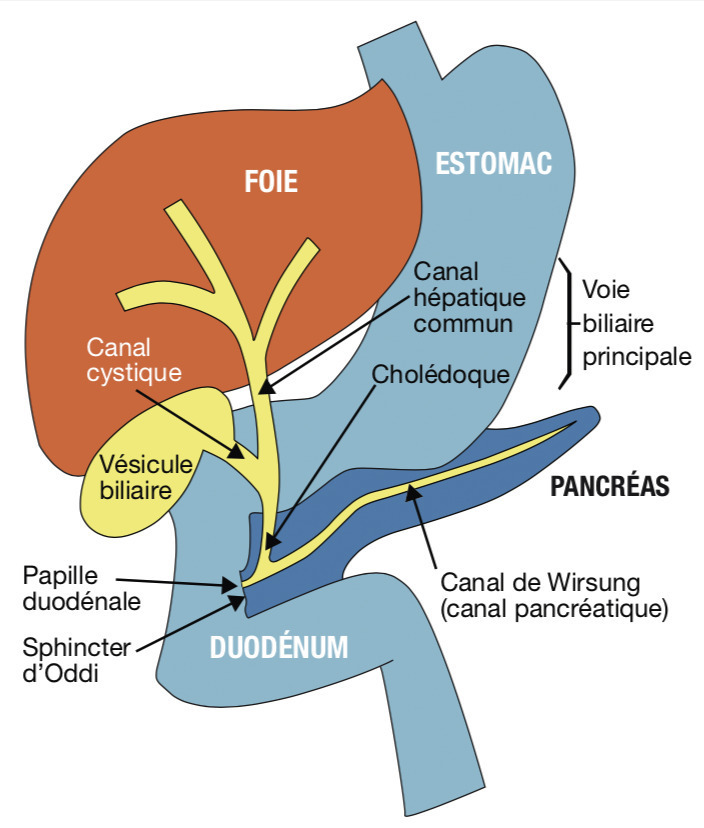
Lorsqu’il existe un obstacle cholédocien en aval de la convergence du canal cystique et du canal hépatique commun, la vésicule est palpée (
Bilan complémentaire
Les phosphatases alcalines (PAL) et les gammaglutamyl-transférases (GGT) sont élevées.
La réalisation d’une échographie abdominale est indispensable.
En cas de dilatation des voies biliaires, d’autres explorations (écho-endoscopie, bili-IRM, voire cholangiographie rétrograde endoscopique) précisent le siège de l’obstacle et permettent de faire des biopsies diagnostiques (lésion pancréatique, adénopathies…), voire des gestes thérapeutiques (sphinctérotomie devant une lithiase de la voie biliaire principale, ampullectomie, etc.).
La dilatation des voies biliaires peut manquer lorsque l’obstacle est récent, les voies biliaires fibreuses ou le parenchyme hépatique cirrhotique (perte d’élasticité). En l’absence de dilatation des voies biliaires, il faut donc interroger le patient sur sa consommation de boissons alcoolisées et de médicaments, rechercher des anticorps anti-tissus (antimitochondries M2, en particulier), faire des sérologies virales.
Causes
Les causes sont classées en fonction de la présence, ou non, d’un obstacle sur les voies biliaires.
Avec obstacle sur les voies biliaires
Les causes diffèrent selon la localisation de l’atteinte.
L’atteinte des gros canaux biliaires peut être liée à trois types d’obstacles :
– obstacle pancréatique : cancer, pancréatite chronique ;
– obstacle papillaire : ampullome (tumeur de l’ampoule de Vater ou papille duodénale,
– obstacle sur les voies biliaires : lithiase, cancer de la voie biliaire principale, sténose postopératoire, compression par une adénopathie ou une tumeur de voisinage, parasite (douves, ascaris).
Les causes d’atteinte des canaux biliaires de petit et moyen calibres sont plus variées :
– cholangite biliaire primitive à un stade tardif. D’origine auto-immune, elle peut évoluer vers une cirrhose. La présence d’anticorps antimitochondries de type M2 oriente le diagnostic ;
– cholangite d’origine médicamenteuse (acide clavulanique associé à l’amoxicilline, macrolides, etc.) ;
– cholangite sclérosante primitive caractérisée par des sténoses des voies biliaires intra- et/ou extrahépatiques à l’origine d’épisodes d’angiocholite et pouvant évoluer vers une cirrhose. Les maladies inflammatoires intestinales (maladie de Crohn et rectocolite hémorragique) y sont fréquemment associées ;
– mucoviscidose et anomalies du transporteur des phospholipides (MDR3) peuvent conduire à des obstacles biliaires (présence de mucus, de microcalculs) ;
– pathologie hépatique infiltrative à l’origine de compression des canaux biliaires (granulomatose, lymphome).
Sans obstacle sur les voies biliaires
Deux grands types de cause liée à des anomalies des transporteurs biliaires (des acides biliaires ou d’autres constituants de la bile) se distinguent ici :
– cholestase récurrente bénigne (épisodes de cholestase récidivants et spontanément résolutifs) ; cholestase gravidique (apparaissant au troisième trimestre de la grossesse et disparaissant après l’accouchement) ;
– cholestase cytokinique liée à une inhibition du transporteur des acides biliaires par des interleukines (IL-1, IL-6…), observée dans des sepsis graves bactériens (pyélonéphrite, pneumonie, péritonite, leptospirose, fièvre typhoïde…) ou dans des hépatites (alcooliques, virales) ; cholestase liée au syndrome inflammatoire de certaines maladies malignes (syndrome d’activation macrophagique au cours du lymphome) et au syndrome paranéoplasique.
Si aucune cause n’est trouvée, une biopsie hépatique doit être pratiquée.
L’auteur déclare n’avoir aucun lien d’intérêts.
* Destruction des hématies dans la moelle osseuse.
Brugel M, Thiefin G. Item 275. Ictère. Rev Prat 2018;68(6):e255-63.
Fargo MV, Grogan SP, Saguil A. Evaluation of jaundice in adults. Am Fam Physician 2017;95(3):164-8.
Gazzin S, Masutti F, Vitek L, Tiribelli A. The molecular basis of jaundice: An old symptom revisited. Liver Int 2017;37(8):1094-102.