Un homme de 19 ans se présente en visite d’expertise médicale initiale à Faaa sur l’île de Tahiti, en Polynésie française. Il n’a pas d’antécédent personnel ou familial. Il n’a aucune plainte fonctionnelle et envisage une carrière au sein du bataillon de marins-pompiers de Marseille. Il est à jour des vaccinations et ne rapporte aucune consommation de toxiques. Son indice de masse corporelle est évalué à 27 kg/m², sans notion d’évolution récente du poids. Sa pression artérielle et sa fréquence cardiaque sont normales. Aucun déficit visuel ou auditif n’est mis en évidence par les tests réalisés à l’antenne. Le tracé électrocardiographique et la bandelette urinaire sont normaux.
L’examen clinique révèle une masse sous-mandibulaire droite isolée supracentimétrique, non adhérente à la peau, fixe et rénitente, que le patient n’avait pas remarquée.
Une échographie cervicale met en évidence un « processus tissulaire à hauteur de la bifurcation carotidienne droite dont les caractères font suspecter un éventuel chémodectome ou paragangliome du corpuscule carotidien ».
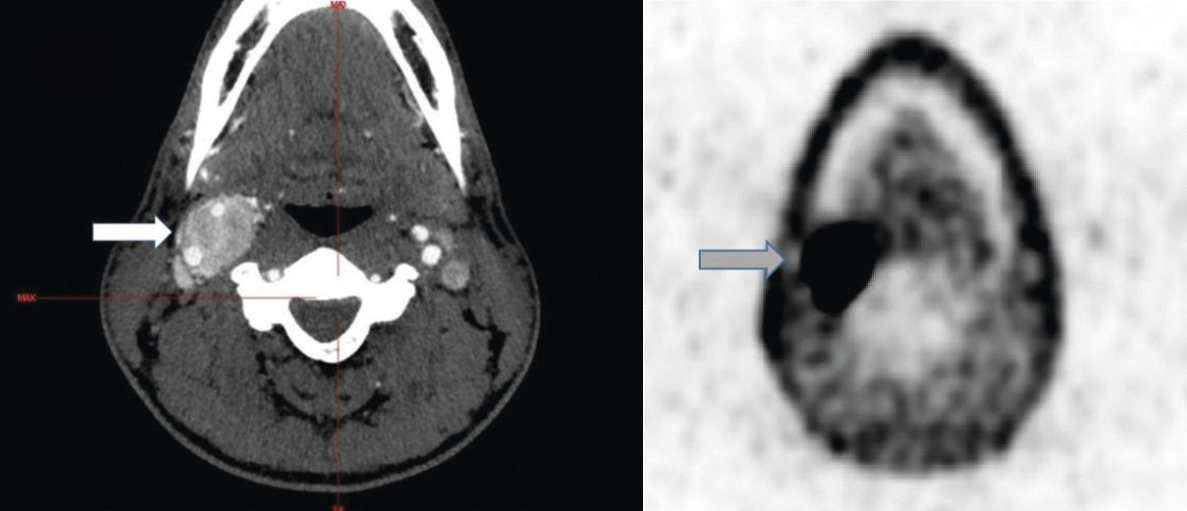
Un scanner cervical apporte confirmation et précise les éléments suivants : lésion de grand axe longitudinal supérieur à 4 cm, de dimensions transversales maximales voisines de 2 à 3 cm, retentissement sur la bifurcation carotidienne avec un écartement de l’origine des carotides externe et interne et un déplacement antérieur de la carotide externe, petites hypodensités centrales pouvant correspondre à un contingent nécrotique, sans lésion controlatérale (fig. A).
Le jeune patient est orienté vers un oto-rhino-laryngologiste à Tahiti, lequel organise une évacuation vers un centre de référence en métropole pour la suite de la prise en charge.
Sur le plan biologique, les dérivés méthoxylés plasmatiques sont normaux (métanéphrine libre plasmatique à 0,17 nmol/L [normale < 0,4] et normétanéphrine libre plasmatique à 0,53 mmol/L [normale < 1,1]). Sur le scanner thoraco-abdomino-pelvien, aucune anomalie n’est décelée, notamment ni lésion hypervasculaire ni atteinte des glandes surrénales. L’imagerie par résonance magnétique (IRM) et l’angio-IRM cervicales objectivent un paragangliome carotidien droit unique de 39 × 23 mm. En TEP-scan à la DOPA, un hypermétabolisme intense est relevé au sein de la lésion, sans autre foyer de fixation suspect (fig. B).
Après signature d’un consentement éclairé, l’étude génétique révèle un variant du gène SDHD à l’état hétérozygote, sous la forme d’une délétion de deux paires de bases entraînant un décalage du cadre de lecture à partir du codon 64 et introduisant un codon stop quatre acides aminés en aval. Ce variant a été considéré comme pathogène et responsable de la symptomatologie du patient. Les mutations au sein du gène SDHD sont transmises de façon dominante, correspondant à un risque de maladie de 50 % pour la descendance. Une étude génétique a été proposée dans le cadre d’un dépistage familial chez les apparentés au premier degré.
Au total, il s’agit d’un paragangliome carotidien droit non sécrétant.
Après réunion de concertation pluridisciplinaire, cette tumeur du glomus carotidien droit a été réséquée, avec pontage carotidien en veine saphène interne homolatérale inversée. Les suites opératoires ont été marquées par une discrète dysphagie aux solides non invalidante, ainsi qu’un syndrome de Claude-Bernard-Horner sur irritation sympathique, en régression. À l’écho-Doppler de contrôle, le pontage carotidien était perméable et la carotide interne en aval de bon flux. Le jeune patient a quitté le service hospitalier sous traitement antalgique et antiagrégant plaquettaire (aspirine 75 mg/j). La réévaluation clinique et l’écho-Doppler de contrôle ont été planifiés trois mois plus tard à Tahiti. Une surveillance annuelle par écho-Doppler est recommandée pour les trois années à venir.
L’aptitude à devenir militaire est peu probable en ces circonstances, même après plusieurs années de recul.
Discussion
Le diagnostic de paragangliome carotidien a donc été posé. Cette tumeur rare est le plus souvent bénigne et les modalités de sa prise en charge sont bien établies.1
À propos du contexte
Si le contexte fortuit de diagnostic est habituel, la résidence du patient en Polynésie a pu compliquer le parcours de soins.
Particularités de la visite d’expertise médicale initiale
La visite d’expertise médicale initiale a pour objectif de rechercher toute lésion ou pathologie susceptible de rendre la personne médicalement inapte à son projet d’engagement militaire. Elle repose sur un interrogatoire (aidé d’un questionnaire complété par le candidat), un examen clinique complet et quelques examens complémentaires.
En Polynésie, les causes d’inaptitude les plus fréquentes sont la consommation de cannabis, l’obésité et le déficit auditif. Il peut arriver que la découverte d’un élément non connu pose l’indication d’examens complémentaires afin d’en préciser le diagnostic et d’évaluer l’aptitude à l’engagement, selon l’arrêté relatif à la détermination du profil médical d’aptitude en cas de pathologie médicale ou chirurgicale.2
La découverte d’une masse cervicale n’est pas inhabituelle. Ses caractéristiques morphologiques et le contexte clinique général permettent le plus souvent une orientation étiologique rapide. Au moindre doute, une échographie cervicale et un contrôle de l’hémogramme permettent de confirmer, le plus souvent, l’aptitude à servir.
Découverte d’une tumeur rare et grave en outre-mer
Organiser une évacuation sanitaire est complexe et trouver le financement pour un accompagnant parmi les proches n’est pas aisé.
Par ailleurs, en Polynésie, des difficultés économiques et logistiques peuvent rendre le recours au médecin aléatoire. L’opportunité d’un examen clinique complet a donc ici été appréciée.
Tumeur endocrine le plus souvent bénigne
Les paragangliomes ou chémodectomes du corpuscule carotidien sont des tumeurs développées aux dépens des chémorécepteurs de la bifurcation carotidienne. Ils se présentent sous la forme d’une masse cervicale, de nature bénigne dans la majorité des cas.3 Il s’agit de tumeurs endocrines provenant du système paraganglionnaire, lui-même constitué de cellules neuroépithéliales dérivant de la crête neurale sur le plan embryologique et dont le cytoplasme possède des granules sécrétoires de catécholamines. Les paragangliomes peuvent se développer le long des chaînes ganglionnaires parasympathiques (région cervicale et base du crâne) et orthosympathiques (région cervicale, le long de la colonne vertébrale et du tractus génito-urinaire). Enfin, ces tumeurs peuvent se développer au niveau des glandes surrénales, il s’agit dans ce cas de phéochromocytomes.3
Rare et concernant l’adulte
Les paragangliomes représentent 0,03 % de l’ensemble des tumeurs du corps humain. Celui du corpuscule carotidien correspond à 0,6 % des paragangliomes, avec une discrète prédominance féminine (4 femmes/3 hommes) et un âge moyen de survenue de 52 ans.3 L’incidence des phéochromocytomes est de 0,47/100 000 personnes-années et celle des paragangliomes de 0,11/100 000 personnes-années. Leur prévalence au sein d’une population de patients hypertendus est évaluée entre 0,1 et 0,6 %.1 Ces tumeurs sont familiales, de transmission autosomique dominante, dans 40 % des cas.1 Le paragangliome cervical est fonctionnel dans 10 % des cas.1 Cette tumeur est considérée comme très rare chez les enfants et les adolescents, avec seulement 23 cas rapportés dans la littérature récente.4
Circonstances de découverte
La présentation clinique du paragangliome peut correspondre à différentes situations. La présence d’une masse cervicale latérale semble être la plus fréquente.5 Elle peut d’ailleurs être faussement rassurante et orienter vers un kyste de l’arc branchial, occasionnant un retard de prise en charge.4
Il peut exister également des signes cliniques en rapport avec la compression locale tumorale : dysphonie, odynophagie, douleur cervicale, vertiges.3
Les paragangliomes cervicaux sont exceptionnellement sécrétants (10 % des cas1). S’ils le sont, il peut exister des crises paroxystiques d’hypertension artérielle (HTA) associées à des céphalées, sueurs, palpitations, nausées,6 ou encore une HTA chronique difficile à équilibrer et accompagnée de douleurs thoraciques ou abdominales.
Autre circonstance diagnostique, il peut arriver que la tumeur soit découverte fortuitement, lors d’un examen d’imagerie ou d’une intervention chirurgicale, en particulier les paragangliomes du thorax, abdominaux ou pelviens.
Enfin, le diagnostic peut être posé au cours d’une recherche génétique dans le cadre d’un dépistage familial.
Prise en charge d’un paragangliome
Le mode de diagnostic et la prise en charge de cette tumeur ont été décrits dans le protocole national de diagnostic et de soins (PNDS) en septembre 2021.1
Démarche diagnostique
Outre un kyste de l’arc branchial, le diagnostic différentiel peut être celui d’une adénopathie cervicale (par exemple dans le cadre d’une mononucléose infectieuse), en particulier si le patient est reçu en consultation dans le cadre d’un syndrome infectieux.4
Imagerie, échographie puis IRM6 redressent alors le diagnostic. Le PNDS recommande de réaliser une imagerie fonctionnelle à l’aide d’analogues de la somatostatine marqués, ces lésions neuro-endocrines comportant des récepteurs SST2 à la somatostatine.1 Ainsi, le scanner couplé à une tomographie par émission de positons (TEP-TDM) marqué au 68Ga-DOTATOC peut être réalisé ; en cas d’indisponibilité de ce traceur, une TEP 18F-FDOPA doit être proposée. Ces examens permettent en outre de rechercher d’autres localisations.
Les dosages des métanéphrines affirment la nature sécrétante ou non de la tumeur. Le dosage actuel des métanéphrines libres plasmatiques ou urinaires est très fiable.1
La confirmation diagnostique formelle ne peut être qu’anatomopathologique. Or la biopsie n’est pas envisageable en routine. Le diagnostic histologique n’est effectué qu’après le retrait chirurgical de la tumeur.
Évaluation de la gravité
En amont du geste chirurgical, il est nécessaire d’établir le diagnostic de gravité du paragangliome en précisant sa taille et le niveau d’envahissement des structures vasculaires de voisinage (bifurcation carotidienne).
La classification de Shamblin établit ainsi une approche de la tumeur à opérer selon trois stades :7
– I, tumeur localisée entre la carotide interne et la carotide externe, facile à réséquer ;
– II, tumeur adhérant à une carotide ou l’entourant partiellement ;
– III, tumeur entourant ou engainant les vaisseaux ; le sous-stade IIIa concerne les tumeurs sans contact avec la base du crâne et le sous-stade IIIb celles qui sont en contact avec la base du crâne et ne laissant pas de segment de carotide accessible.
Dans le cas présenté, la bifurcation carotidienne ainsi que la carotide externe étaient incluses dans la tumeur, évoquant un stade IIIa selon la classification de Shamblin.
Le traitement chirurgical comporte un risque vasculaire croissant du stade I au stade III. Plus le diagnostic est tardif, plus grand est le risque que la tumeur se développe aux dépens des carotides et des autres structures de voisinage, rendant son ablation difficile et nécessitant une reconstruction.
Recherche de lésions associées
La recherche d’autres localisations est également nécessaire, notamment en cas de mutation génétique.
Une mutation du gène SDHD peut être associée à un phéochoromocytome (10 à 25 % des cas), à un paragangliome thoraco-abdominal (20 à 25 % des cas) et à un autre paragangliome de la tête et du cou (85 % des cas). Signalons enfin l’association possible avec des cancers du rein ainsi qu’avec des tumeurs stromales gastro-intestinales ou à des adénomes hypophysaires.8
Traitement optimal
L’enjeu de la prise en charge est le contrôle tumoral. Il peut être obtenu par des traitements locaux (chirurgie, radiologie interventionnelle, radiofréquence ou cryothérapie, radiothérapie externe) ou généraux (radiothérapie métabolique, chimiothérapie, antiangiogéniques). Une embolisation préchirurgicale peut également être indiquée, mais son utilisation est discutée.
En cas de tumeur sécrétante, la chirurgie nécessite une préparation, en raison des difficultés à maintenir une hémodynamique stable (libération hormonale par manipulation de la tumeur avec variations de la fréquence cardiaque et de la pression artérielle).1, 9
La lenteur d’évolution de certaines tumeurs et les effets secondaires des traitements peuvent enfin plaider pour une surveillance seule par examens d’imagerie répétés périodiquement.
Complications chirurgicales
Le décès, l’accident vasculaire cérébral et la lésion d’un nerf crânien sont à redouter.10
Enquête génétique
Le diagnostic génétique permet le dépistage familial de la maladie et un conseil pour la filiation. Un grand nombre de mutations, germinales ou somatiques peut se rencontrer.11 Le PNDS recommande de rechercher systématiquement dix mutations au sein d’un panel d’au moins dix gènes par séquençage de nouvelle génération.1
Dans le cas présenté, le dépistage familial permettrait la découverte précoce de tumeurs de plus petite taille, à faible risque de complications1, 7, chez des apparentés au premier degré.
Quelle évolution ?
La fréquence des récidives n’est pas clairement connue, mais un suivi de cohorte de 39 cas a mis en évidence 6 récidives.12 La surveillance recommandée se poursuit à vie (IRM tous les 2 ans).
Le patient a donné son consentement pour la rédaction de cet article.
1. Protocole national de diagnostic et de soins pour le phéochromocytome et le paragangliome, https://www.has-sante.fr/upload/docs/application/pdf/2021-10/pheochromocytomes_et_paragangliomes_pnds.pdf
2. Arrêté du 26 novembre 2021 modifiant l’arrêté du 29 mars 2021 relatif à la détermination du profil médical d’aptitude en cas de pathologie médicale ou chirurgicale.
3. Bougrine F, Maamouri F, Doghri R, Msakni I, Sabbegh Znaidi N, Chouchane O, et al. Une tumeur rare du glomus carotidien. Journal des maladies vasculaires 2008;33:214-7.
4. Kotsis T, Christoforou P, Nastos C. Carotid paraganglioma in adolescence – Clinical picture – Surgical technique and review of the litterature. Case Reports in Vascular Medicine, Volume 2019, Article ID 6182783.
5. Pitchaiprasert S, Boonchaya-Anant P, Snaboon T. Carotid body paraganglioma. Clin Case Rep 2019;00:1-2.
6. Abdelhady K, Durgam S, Orza D, Massad MG. Left atrial and carotid body paraganglioma. Ann Thorac Surg 2017;103:323-5.
7. Lamblin E, Atallah I, Reyt E, Schmerber S, Magne JL, Righini CA. Neurovascular complications following carotid body paraganglioma resection. European Annals of Otorhinolaryngology. Head and Neck Diseases 2016;133:319-24.
8. Benn DE, Robinson BG, Clifton-Bligh RJ. Fifteen years of paraganglioma, clinical manifestations of paragnaglioma syndromes types 1-5. Endocrine-Related Cancer 2015;22:T91-T103.
9. Deljou A, Kohlenberg JD, Weingarten TN, Bancos I, Young WF Jr, Schroeder DR, et al. Hemodynamic instability during percutaneous ablation of extra-adrenal metastases of pheochromocytoma and paragangliomas: a case series. BMC Anesthesiol 2018;18:158.
10. Robertson V, Poli F, Saratzis A, Ross Naylor A. A systematic review and meta-analysis of the presentation and surgical management of patients with carotid body tumours. Eur J Vasc Endovasc Surg 2019;57:477-86.
11. Gieldon L, William D, Hackmann K, Jahn W, Jahn A, Wagner J, et al. Optimizing genetic workup in pheochromocytoma and paraganglioma by integrating diagnostic and research approaches. Cancers 2019;11:809.
12. Lamblin A, Pigny P, Tex G, Rouaix-Emery N, Porchet N, Leteurtre E, et al. Paragangliomes : profil clinique et sécrétoire, à propos de 39 cas. Annales de chirurgie 2005;130:157-61.