Les circonstances de découverte fortuite d’une maladie kystique rénale sont variées : hématurie, infection urinaire, colique néphrétique, douleur rénale, hypertension artérielle (HTA), insuffisance rénale chronique, grossesse, examen clinique de l’abdomen, échographie, scanner ou imagerie par résonance magnétique (IRM) pour une autre raison, etc. Elle peut être découverte à tout âge.
Les premiers diagnostics évoqués par le radiologue sont en général la multikystose simple ou la polykystose rénale autosomique dominante (PKRAD). Cependant, plusieurs autres causes peuvent être à l’origine d’une maladie kystique rénale. Un interrogatoire minutieux permet de préciser le diagnostic étiologique et d’évaluer le retentissement de la maladie. Le cas du monokyste simple n’est pas évoqué ici, puisqu’il ne nécessite pas de prise en charge particulière.
Rechercher des signes associés
Les néphropathies chroniques – notamment kystiques – sont la première cause d’HTA secondaire. Il est donc nécessaire, en premier lieu, de mesurer la pression artérielle du patient. Il est également indiqué de rechercher une insuffisance rénale chronique (mesure de la créatininémie et estimation du débit de filtration glomérulaire [DFG]), une protéinurie (habituellement absente ou de faible débit dans les maladies rénales kystiques) et des anomalies urologiques favorisées par les kystes (douleurs rénales aiguës ou chroniques, hématuries, infections urinaires, lithiases urinaires).
Caractériser les kystes rénaux
Il est essentiel de préciser la taille des reins, le nombre de kystes et leur taille, leur répartition uni- ou bilatérale, leur localisation (corticale, médullaire, diffuse ou parfois sinusale) et leur morphologie.
La classification morphologique de Bosniak permet de décrire les kystes rénaux selon leur potentiel de malignité. Initialement décrite en tomodensitométrie (TDM), elle a ensuite été adaptée à l’échographie et à l’IRM ; elle distingue quatre variétés de kystes :
– kystes simples (Bosniak de type 1), de loin les plus fréquents, pour lesquels une échographie peut suffire ;
– kystes Bosniak de type 2,3 et 4, pour lesquels la TDM ou l’IRM est utile.
Certains kystes de type 2 sont à surveiller par imagerie. Les kystes de type 3 et 4 sont suspects d’être tumoraux et nécessitent un avis urologique.1
Au-delà de la classification de Bosniak, plusieurs repères sont utiles2 (
Si les deux reins sont de taille augmentée, avec des contours bosselés et des kystes innombrables, corticaux et médullaires, le diagnostic de PKRAD est hautement probable.
Un nombre limité de kystes rénaux, sans néphromégalie, peut également faire évoquer une PKRAD. Il peut s’agir d’une forme de l’adolescent ou de l’adulte jeune, ou d’une polykystose de type 2 (PKD2) chez un patient plus âgé.
Lorsqu’ils sont associés à des angiomyolipomes rénaux, le diagnostic de sclérose tubéreuse de Bourneville – maladie génétique neurocutanée rare caractérisée par des hamartomes multisystèmes – est possible.
En cas de kystes rénaux associés à des tumeurs rénales solides, la maladie de von Hippel-Lindau, syndrome familial de prédisposition aux cancers, doit être évoquée.
Chez l’enfant, les kystes rénaux purement médullaires (ne bosselant pas les contours rénaux) peuvent faire penser à une polykystose récessive ou à une néphronophtise. En revanche, chez l’adulte, il peut s’agir d’une néphropathie tubulo-interstitielle chronique héréditaire autosomique dominante ou acquise.
Les kystes rénaux sinusaux (développés dans le sinus rénal, en dehors du parenchyme) suggèrent, quant à eux, un kyste parapyélique (habituellement unique) ou des kystes péripyéliques d’origine lymphatique, bilatéraux (ils s’insinuent entre les tiges calicielles et peuvent donner une fausse impression d’hydronéphrose obstructive en échographie ou sur un scanner non injecté).
Enfin, les kystes rénaux unilatéraux – rares et insolites – peuvent être liés à une dysplasie rénale multikystique (chez l’enfant), à une anomalie génétique (mutation du gène HNF1ß [hepatocyte nuclear factor 1 beta] et mosaïque PKD [polycystic kidney disease]), à une maladie infectieuse (parasitaire ou bactérienne) ou à une tumeur (bénigne ou maligne).
Existe-t-il des anomalies extrarénales associées ?
Plusieurs anomalies extrarénales sont susceptibles d’être associées aux maladies kystiques rénales (
Kystes hépatiques
La présence simultanée de kystes rénaux et hépatiques rend le diagnostic de PKRAD hautement probable. En revanche, l’absence de kystes hépatiques à l’échographie n’écarte pas ce diagnostic. L’âge du patient (la prévalence des kystes hépatiques dans la PKRAD augmente progressivement avec l’âge), son sexe (les kystes hépatiques sont globalement plus nombreux et volumineux chez les femmes) ainsi que la technique d’imagerie employée (l’IRM est plus sensible que l’échographie pour détecter les kystes) sont autant de paramètres à prendre en compte.
Diabète
L’association des kystes rénaux avec un diabète fait évoquer le diagnostic de mutation du gène HNF1ß.3 Cette affection héréditaire autosomique dominante est la deuxième cause de néphropathie kystique de l’adulte. Le diabète, de type MODY 5, peut apparaître tardivement, voire jamais. D’autres anomalies fréquemment associées (hypertransaminasémie, hypomagnésémie, hyperurémie goutteuse, utérus bicorne, malformation urinaire) peuvent également suggérer ce diagnostic.
Enquête familiale : indispensable
L’interrogatoire du patient sur les antécédents familiaux, en matière de kystes rénaux, problèmes urologiques, HTA, insuffisance rénale, etc. permet d’ébaucher un arbre généalogique.
Dans le cas où l’un des parents du patient est atteint de PKRAD, une simple échographie révélant la présence de plus de trois kystes rénaux (avant 40 ans) ou de plus de deux kystes par rein (après 40 ans) permet d’affirmer le diagnostic de PKRAD.2
Que faire en cas d’interrogatoire familial négatif ?
Même si l’interrogatoire familial est négatif, une PKRAD peut être présente chez des apparentés qui l’ignorent. Le patient doit alors informer les membres de sa famille proche, et convaincre de la nécessité de réaliser une échographie des reins et du foie, au moins chez ses deux parents, puis éventuellement chez ses frères et sœurs ainsi que chez ses enfants.
Pour autant, une enquête échographique familiale négative n’exclut pas le diagnostic de polykystose dominante : en effet, le patient peut avoir une néomutation (environ 5 % des PKRAD).
Les mutations de HNF1ß sont aussi à l’origine de néphropathie kystique autosomique dominante avec ou sans diabète.
Maladies rénales récessives dépistées dès l’enfance
Les principales maladies rénales kystiques récessives (polykystose rénale récessive, néphronopthise) sont le plus souvent révélées dès l’enfance ; leur survenue est plus fréquente en cas de consanguinité. Plusieurs cas sont possibles dans la fratrie, mais les parents ne développent généralement pas de kystes rénaux.4
Test génétique : quand l’envisager et quelle prise en charge ?
Il est possible de diagnostiquer une maladie polykystique rénale, dès le plus jeune âge, par une simple prise de sang. Pour l’enfant à naître, l’analyse d’un prélèvement du placenta ou du liquide amniotique peut amener au diagnostic.
Le recours au séquençage en milieu spécialisé pour identifier la cause d’une maladie rénale kystique est le plus souvent proposé dans deux circonstances (
L’identification d’une mutation du gène PKD2 (environ 15 % des cas) est, par exemple, généralement de bon pronostic, car elle est associée à une survie rénale médiane d’environ quatre-vingts ans. En revanche, l’identification d’une mutation du gène PKD1 (80 % des cas) est associée à une survie rénale médiane d’environ soixante ans, ce qui invite à mettre en place sans délai toutes les mesures de protection rénale.
PKRAD : protéger pour limiter les complications
Les objectifs principaux face à la découverte d’une PKRAD sont de ralentir la progression de la maladie rénale et de prendre en charge d’éventuelles complications extrarénales.7
Baisser la pression artérielle
Chez les patients jeunes (18 à 50 ans), dont le DFG est supérieur à 60 mL/min/1,73 m², la pression artérielle doit être maintenue en-dessous de 110/75 mmHg. Dans tous les autres cas, une pression artérielle inférieure à 130/80 mmHg est préconisée.
Mesures hygiénodiététiques
Il y a lieu de réduire les apports sodés à moins de 6 g/j, de maintenir un indice de masse corporelle (IMC) à moins de 24 et d’augmenter le volume de boissons à plus de 3 L/j.
Tolvaptan : protection rénale partielle, risque hépatique
Le tolvaptan, antagoniste oral des récepteurs V2 à la vasopressine, est connu pour ralentir la croissance des kystes et le déclin de la fonction rénale. C’est un traitement d’exception ; les patients doivent être éligibles (âge, volume rénal, éventuelles complications urologiques et évolution de lafonction rénale au fil des ans comptent parmi les critères de sélection) mais aussi motivés car ce traitement induit un syndrome polyuropolydipsique intense. De plus, le patient doit se soumettre à une surveillance hépatique mensuelle sur une période de dix-huit mois du fait d’un risque d’hépatite médicamenteuse.7
Surveiller pour limiter les complications extrarénales
Le dépistage, tous les cinq à dix ans, par angio-IRM cérébrale (ou angioscanner) d’un éventuel anévrisme intracrânien est à mettre en place.8
En cas d’hépatomégalies kystiques massives – qui surviennent principalement chez les femmes et concernent environ 10 % d’entre elles –, il faut éviter les œstrogènes, et discuter les options thérapeutiques suivantes : analogues de la somatostatine, radiologie interventionnelle et chirurgie.
Quand le contexte oriente la prise en charge : trois cas particuliers
• Si l’échographie anténatale révèle une hyperéchogénicité de la médullaire rénale et/ou des kystes rénaux et/ou une néphromégalie, trois diagnostics principaux sont évoqués : polykystose dominante, polykystose récessive, mutations de HNF1ß.5 Une prise en charge en néphrologie pédiatrique et un conseil génétique doivent être proposés.
• Après 60 ans, il est possible d’observer, chez des sujets en bonne santé, un nombre limité de kystes rénaux (habituellement moins de quatre par rein). Pression artérielle et fonction rénale sont le plus souvent normales. La multikystose rénale simple est alors évoquée.6 Si les kystes apparaissent bénins à l’imagerie, aucune surveillance ultérieure n’est nécessaire. En cas d’hypertension artérielle et/ou d’insuffisance rénale chronique, une PKRAD de type 2 peut être discutée.
• Enfin, il est banal de voir se développer des kystes rénaux chez les patients dialysés.
Que dire à vos patients ?
Chez le sujet âgé, la multikystose est une anomalie fréquente et bénigne qui ne nécessite, le plus souvent, qu’une simple surveillance de la pression artérielle et de la fonction rénale.
Pour le sujet jeune, la PKRAD est plus probable et doit orienter vers un avis spécialisé.
Il est possible de se rapprocher des associations existantes :
https://www.polykystose.org/ et www.airg-france.fr (remarquable livret d’informations sur la PKRAD, téléchargeable).
1. Pachev A, De Kerviler É. Kystes rénaux : lesquels sont suspects ? Rev Prat Med Gen 2021;35(1054);73-4.
2. Pei Y, Watnick T. Diagnosis and Screening of Autosomal Dominant Polycystic Kidney Disease. Adv Chronic Kidney Dis 2010;17(2):140–52.
3. Haute Autorité de santé. Protocole national de diagnostic et de soins (PNDS). Maladie liée à HNF1 béta. Mars 2020.
4. Kurschat CE, Müller RU, Franke M, et al. An approach to cystic kidney diseases: the clinician’s view. Nat Rev Nephrol 2014;10(12):687-99.
5. Gimpel C, Avni FE, Bergmann C, et al. Perinatal Diagnosis, Management, and Follow-up of Cystic Renal Diseases: A Clinical Practice Recommendation With Systematic Literature Reviews. JAMA Pediatr 2018;172(1):74-86.
6. Simms RJ, Ong ACM. How simple are ‘simple renal cysts’? Nephrol Dial Transplant 2014;29(Suppl4):iv106–iv112.
7. Chebib FT, Perrone RD, Chapman AB, et al. A Practical Guide for Treatment of Rapidly Progressive ADPKD with Tolvaptan. J Am Soc Nephrol 2018;29(10):2458-70.
8. Flahault A, Trystram D, Nataf F, et al. Screening for intracranial aneurysms in autosomal dominant polycystic kidney disease is cost-effective. Kidney Int 2018;93(3):716-26.
Dans cet article
 Encadrés
Encadrés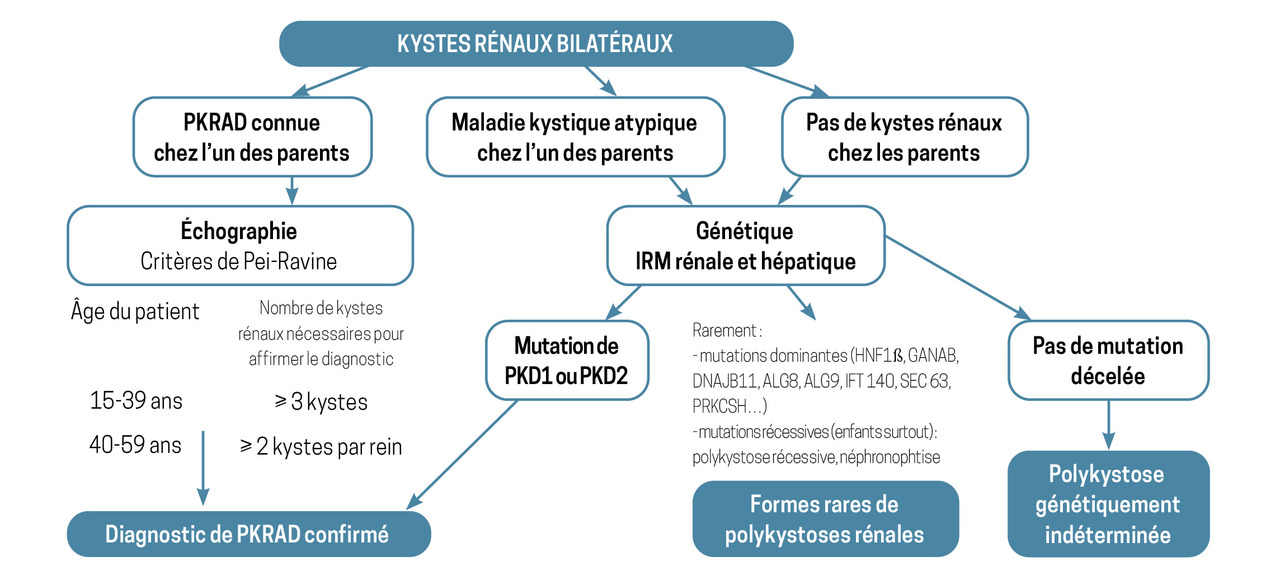

La découverte de kystes rénaux multiples doit faire rechercher des signes associés (HTA, insuffisance rénale, douleurs, complications urologiques…).
Plusieurs éléments plaident en faveur d’une polykystose dominante : antécédents familiaux, kystes rénaux innombrables, reins de taille augmentée, kystes hépatiques associés.
Une multikystose simple est plutôt évoquée après 60 ans, si l’on dénombre moins de quatre kystes par rein, en l’absence de kystes hépatiques associés et d’antécédents familiaux et lorsque pression artérielle et fonction rénale sont normales.