Le but du dépistage est d’identifier les enfants atteints d’un syndrome drépanocytaire majeur qui, dès l’âge de 3 mois, peut menacer leur vie et de mettre en place le parcours de soins nécessaire à leur prise en charge.
La drépanocytose est une maladie génétique de transmission autosomique récessive liée à la présence de la mutation bS, responsable de l’expression du variant S de l’hémoglobine (HbS). La maladie ne se manifeste que chez les patients dont la mutation touche les deux allèles du gène ß-globine. Les syndromes drépanocytaires majeurs regroupent les sujets homozygotes avec la mutation bS en double copie (dits homozygotes SS), et les sujets hétérozygotes composites associant la mutation bS avec une mutation différente sur l’autre gène ß-globine (il s’agit le plus souvent de l’hémoglobine C [patients hétérozygotes composites SC] ou d’une b-thalassémie [patients S/b-thalassémiques tableau 1]). Les sujets qui ont la mutation bS à l’état hétérozygote ne sont pas symptomatiques.
Objectifs du dépistage néonatal
En France, la drépanocytose fait partie des cinq maladies dépistées à la naissance, dont la liste est fixée par arrêté ministériel.1
L’objectif principal est de repérer les nouveau-nés atteints de syndromes drépanocytaires majeurs. Les nouveau-nés drépanocytaires sont en bonne santé à la naissance et deviennent symptomatiques lorsque le taux d’hémoglobine fœtale (HbF), protégeant habituellement le nouveau-né, diminue et celui de l’HbS augmente. Les complications de la drépanocytose peuvent survenir dès l’âge de 3 mois et menacer la vie de l’enfant. En 2016, 431 enfants ont été dépistés, soit une incidence de la maladie à la naissance de 1/771 dans la population testée.2
D’autres hémoglobinopathies sévères, telles que les syndromes b-thalassémiques et les hémoglobinoses H, font également partie des maladies dépistées par ce programme. En 2016, 10 syndromes b-thalassémiques ont été repérés.2
Le dépistage néonatal permet également d’identifier les nouveau-nés hétérozygotes. Ces derniers ne sont pas malades et n’ont pas de bénéfice direct à avoir été dépistés. En 2016, 10 160 nouveau-nés hétérozygotes A/S ont été identifiés.
L’objectif principal est de repérer les nouveau-nés atteints de syndromes drépanocytaires majeurs. Les nouveau-nés drépanocytaires sont en bonne santé à la naissance et deviennent symptomatiques lorsque le taux d’hémoglobine fœtale (HbF), protégeant habituellement le nouveau-né, diminue et celui de l’HbS augmente. Les complications de la drépanocytose peuvent survenir dès l’âge de 3 mois et menacer la vie de l’enfant. En 2016, 431 enfants ont été dépistés, soit une incidence de la maladie à la naissance de 1/771 dans la population testée.2
D’autres hémoglobinopathies sévères, telles que les syndromes b-thalassémiques et les hémoglobinoses H, font également partie des maladies dépistées par ce programme. En 2016, 10 syndromes b-thalassémiques ont été repérés.2
Le dépistage néonatal permet également d’identifier les nouveau-nés hétérozygotes. Ces derniers ne sont pas malades et n’ont pas de bénéfice direct à avoir été dépistés. En 2016, 10 160 nouveau-nés hétérozygotes A/S ont été identifiés.
Organisation du dépistage néonatal
L’organisation du dépistage néonatal était confiée jusqu’à fin février 2018 à l’Association française pour le dépistage et la prévention des handicaps de l’enfant (AFDPHE). Débuté par des expériences pilotes en Guadeloupe et en Martinique dès 1981, ce programme est depuis 1992 réalisé chez tous les nouveau-nés des départements et régions d’outre-mer. En métropole, il a été étendu progressivement à partir de 1995, pour atteindre l’ensemble du territoire en 2000. Depuis mars 2018, le dépistage néonatal est géré de manière régionale par les agences régionales de santé, qui délèguent cette mission aux centres régionaux de dépistage néonatal.
Actuellement, le dépistage néonatal de la drépanocytose est systématique pour tous les nouveau-nés des départements, régions et collectivités d’outre-mer, alors qu’il est ciblé en métropole. Le ciblage concerne les nouveau-nés dont les parents appartiennent à un groupe à risque pour la drépanocytose selon des critères définis par l’AFDPHE (tableau 2). En 2016, 39,4 % des naissances ont bénéficié du dépistage de la drépanocytose en métropole. Il existe une importante disparité régionale allant de 9,1 % en Bretagne à 73,6 % en Île-de-France.2 Le ciblage, fondé sur l’origine géographique, est régulièrement remis en question par les professionnels de santé impliqués en raison notamment de la difficulté d’identifier les sujets à risque : imprécision des réponses des couples interrogés sur leurs origines géographiques, brassage des populations, procréations médicalement assistées, adoptions… Une généralisation du dépistage néonatal de la drépanocytose permettrait une simplification du processus, limitant le ressenti de stigmatisation et facilitant la reconnaissance de la drépanocytose comme un problème de santé publique en France.
Actuellement, le dépistage néonatal de la drépanocytose est systématique pour tous les nouveau-nés des départements, régions et collectivités d’outre-mer, alors qu’il est ciblé en métropole. Le ciblage concerne les nouveau-nés dont les parents appartiennent à un groupe à risque pour la drépanocytose selon des critères définis par l’AFDPHE (tableau 2). En 2016, 39,4 % des naissances ont bénéficié du dépistage de la drépanocytose en métropole. Il existe une importante disparité régionale allant de 9,1 % en Bretagne à 73,6 % en Île-de-France.2 Le ciblage, fondé sur l’origine géographique, est régulièrement remis en question par les professionnels de santé impliqués en raison notamment de la difficulté d’identifier les sujets à risque : imprécision des réponses des couples interrogés sur leurs origines géographiques, brassage des populations, procréations médicalement assistées, adoptions… Une généralisation du dépistage néonatal de la drépanocytose permettrait une simplification du processus, limitant le ressenti de stigmatisation et facilitant la reconnaissance de la drépanocytose comme un problème de santé publique en France.
Tests de dépistage
Le dépistage néonatal est réalisé à partir d’un échantillon de sang total obtenu par piqûre au talon du nouveau-né et déposé sur un buvard. Le prélèvement est réalisé à la maternité, après accord des parents, 72 heures après la naissance.
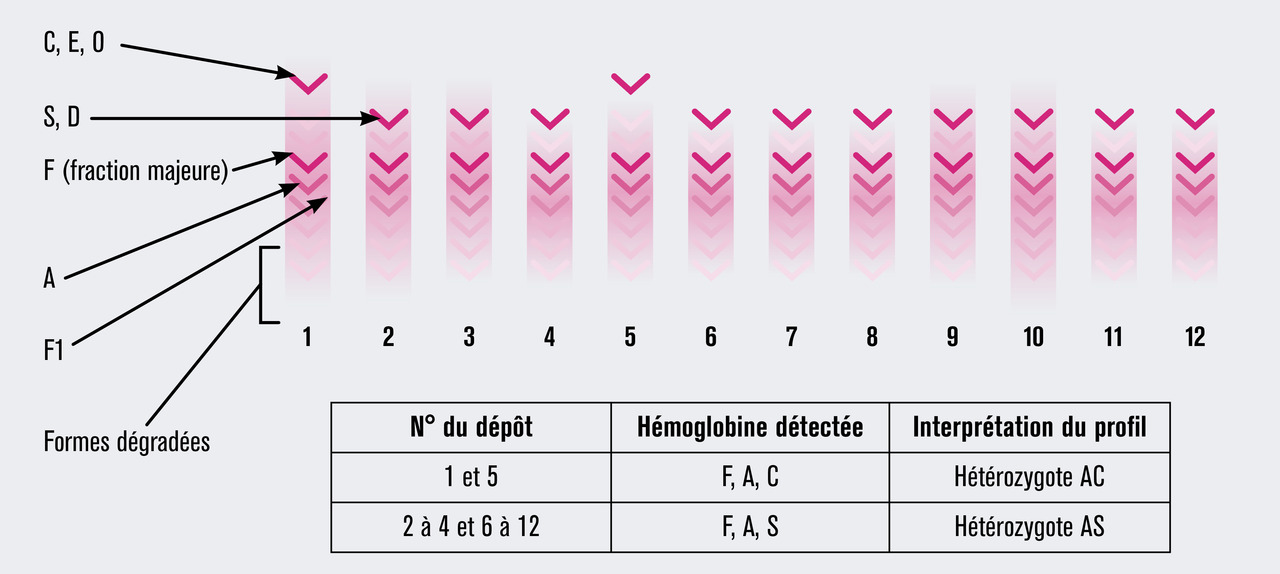
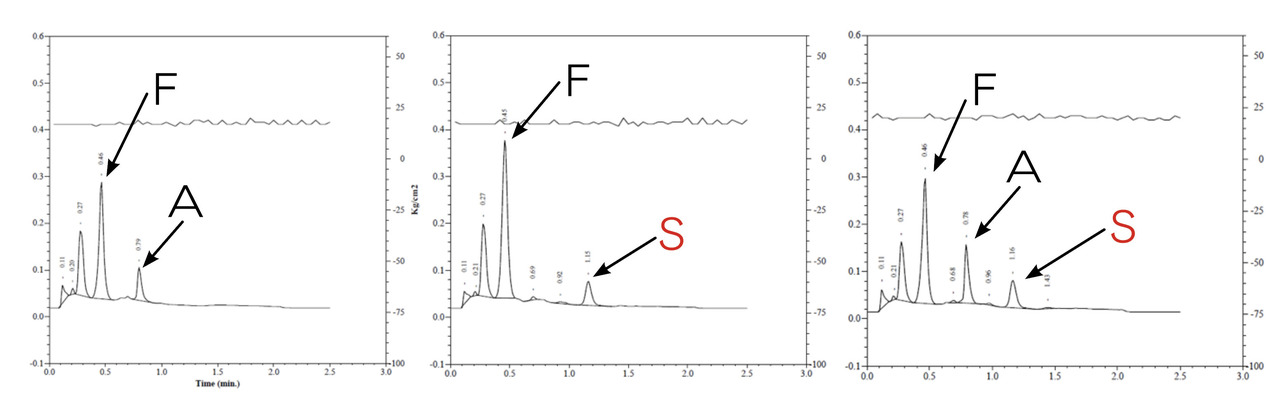
L’analyse est fondée sur des techniques séparatives, électrophorèse et chromatographie, permettant d’une part de séparer les hémoglobines en fonction de leurs caractéristiques physicochimiques et d’autre part de quantifier les différentes fractions d’hémoglobine. Les hémoglobines « anormales », ou variants de l’hémoglobine, sont due à des mutations ponctuelles qui modifient un acide aminé. Pour l’HbS, par exemple, la mutation est responsable du remplacement d’un acide glutamique, acide aminé chargé négativement, par une valine, acide aminé apolaire. Cette modification de charge est à l’origine d’une différence de migration électrophorétique permettant l’identification présomptive du variant (fig. 1). De même, cette modification d’un acide aminé modifie la force d’interaction de l’HbS sur colonne chromatographique, permettant aussi l’identification présomptive du variant en chromatographie liquide haute performance (fig. 2). D’autres techniques font actuellement émergence, notamment la spectrométrie de masse.
Tout résultat positif doit systématiquement être vérifié à l’aide d’une technique différente de celle utilisée en première intention.1 L’utilisation sur un même échantillon d’un second test différent du premier limite le nombre de faux positifs et augmente la spécificité du dépistage.
L’analyse est fondée sur des techniques séparatives, électrophorèse et chromatographie, permettant d’une part de séparer les hémoglobines en fonction de leurs caractéristiques physicochimiques et d’autre part de quantifier les différentes fractions d’hémoglobine. Les hémoglobines « anormales », ou variants de l’hémoglobine, sont due à des mutations ponctuelles qui modifient un acide aminé. Pour l’HbS, par exemple, la mutation est responsable du remplacement d’un acide glutamique, acide aminé chargé négativement, par une valine, acide aminé apolaire. Cette modification de charge est à l’origine d’une différence de migration électrophorétique permettant l’identification présomptive du variant (fig. 1). De même, cette modification d’un acide aminé modifie la force d’interaction de l’HbS sur colonne chromatographique, permettant aussi l’identification présomptive du variant en chromatographie liquide haute performance (fig. 2). D’autres techniques font actuellement émergence, notamment la spectrométrie de masse.
Tout résultat positif doit systématiquement être vérifié à l’aide d’une technique différente de celle utilisée en première intention.1 L’utilisation sur un même échantillon d’un second test différent du premier limite le nombre de faux positifs et augmente la spécificité du dépistage.
Profils identifiés
Le dépistage néonatal identifie les profils suivants :
– les nouveau-nés sans hémoglobinopathie ont une majorité d’HbF et de l’HbA en rapport avec le terme de naissance ;
– les nouveau-nés hétérozygotes AS ont une majorité d’HbF, de l’HbA et de l’HbS avec un taux d’HbA toujours supérieur à l’HbS ;
– les nouveau-nés atteints du syndrome drépanocytaire majeur ont une majorité d’HbF et de l’HbS, associées soit à une absence d’HbA (S/S ou S/b0-thalassémie), soit à de faible quantité d’HbA (S/b+-thalassémie), soit à de l’HbC (S/C), ou de l’HbO-Arab (S/O-Arab)…
Comme les autres dépistages néonataux, le dépistage de la drépanocytose doit faire face aux risques de faux négatifs (oubli de prélèvement…) auquel s’ajoute le défaut de ciblage. En 2016, 4 nouveau-nés atteints de syndromes drépanocytaires majeurs, mais non dépistés, ont été diagnostiqués ultérieurement.3 La transfusion du nouveau-né en culots globulaires constitue un autre risque de faux négatif.
– les nouveau-nés sans hémoglobinopathie ont une majorité d’HbF et de l’HbA en rapport avec le terme de naissance ;
– les nouveau-nés hétérozygotes AS ont une majorité d’HbF, de l’HbA et de l’HbS avec un taux d’HbA toujours supérieur à l’HbS ;
– les nouveau-nés atteints du syndrome drépanocytaire majeur ont une majorité d’HbF et de l’HbS, associées soit à une absence d’HbA (S/S ou S/b0-thalassémie), soit à de faible quantité d’HbA (S/b+-thalassémie), soit à de l’HbC (S/C), ou de l’HbO-Arab (S/O-Arab)…
Comme les autres dépistages néonataux, le dépistage de la drépanocytose doit faire face aux risques de faux négatifs (oubli de prélèvement…) auquel s’ajoute le défaut de ciblage. En 2016, 4 nouveau-nés atteints de syndromes drépanocytaires majeurs, mais non dépistés, ont été diagnostiqués ultérieurement.3 La transfusion du nouveau-né en culots globulaires constitue un autre risque de faux négatif.
Rendu des résultats
Compte tenu du nombre important de nouveau-nés dépistés, les résultats normaux ne sont pas communiqués aux familles. Ces résultats restent disponibles auprès des laboratoires de dépistage et du Centre régional de dépistage néonatal pendant 20 ans.
Les résultats des nouveau-nés hétérozygotes, qui ne sont pas malades, sont rendus aux parents par simple courrier ou au décours d’une consultation selon les organisations locales.
Les résultats des nouveau-nés possiblement atteints d’un syndrome drépanocytaire majeur sont adressés au pédiatre référent avec une fiche d’identification d’un nouveau cas. Celui-ci organise une consultation d’annonce avec les parents et le nouveau-né. Le dépistage néonatal permet de repérer les nouveaux cas, mais une confirmation diagnostique est nécessaire. Cette confirmation a lieu au décours de la première consultation avec réalisation d’un prélèvement veineux chez l’enfant et ses parents.
Les résultats des nouveau-nés hétérozygotes, qui ne sont pas malades, sont rendus aux parents par simple courrier ou au décours d’une consultation selon les organisations locales.
Les résultats des nouveau-nés possiblement atteints d’un syndrome drépanocytaire majeur sont adressés au pédiatre référent avec une fiche d’identification d’un nouveau cas. Celui-ci organise une consultation d’annonce avec les parents et le nouveau-né. Le dépistage néonatal permet de repérer les nouveaux cas, mais une confirmation diagnostique est nécessaire. Cette confirmation a lieu au décours de la première consultation avec réalisation d’un prélèvement veineux chez l’enfant et ses parents.
Vérification par un second test
Un dépistage positif impose une confirmation biologique. Des renseignements complémentaires sont nécessaires à l’interprétation : données hématologiques (résultat de l’hémogramme avec indices érythrocytaires), bilan martial (au minimum la ferritine et la protéine C-réactive), recherche d’une éventuelle transfusion.
Le diagnostic utilise les mêmes techniques séparatives que le dépistage néonatal, à savoir l’électrophorèse et la chromatographie. Tout comme pour le dépistage, l’association de techniques différentes est nécessaire pour permettre l’identification des variants cliniquement significatifs : S, C, D-Punjab, E, O-Arab et Lepore. L’interprétation tient compte de l’évolution physiologique des différentes fractions d’hémoglobine au cours du développement. L’HbF majoritaire à la naissance va décroître et l’HbA va s’accroître pour atteindre des valeurs « adultes », respectivement HbF inférieure à 2 % et HbA égale à 95-98 % entre l’âge de 8 mois à 1 an. La fraction d’HbA2, fraction minoritaire, est difficilement interprétable chez l’enfant de moins de 6 mois ; elle atteint entre 8 mois et 1 an des valeurs « adultes » (HbA2 : 2,2-3,4 %). L’étude de l’hémoglobine des parents est importante pour préciser l’hémoglobinopathie de l’enfant. En effet, à partir de l’étude de l’hémoglobine, il est difficile chez l’enfant de moins de 1 an de faire le diag- nostic différentiel entre les formes S/S, S/b0-thalassémie ou encore S/PHHF (persistance héréditaire de l’hémoglobine F). Le diagnostic différentiel est essentiel car cette dernière association S-PHHF ne constitue pas un syndrome drépanocytaire majeur.
La confirmation diagnostique permet d’éliminer les rares faux positifs générés par le dépistage néonatal. Il s’agit essentiellement d’associations difficiles à dépister à la naissance : S-PHHF, S-Hope, S-K-Woolwich…
Les indications de diagnostic moléculaire sont limitées à la réalisation préalable d’une étude précise de l’hémoglobine. Les syndromes pathologiques sévères qui nécessitent une confirmation moléculaire sont le syndrome drépanocytaire majeur en l’absence d’étude familiale complète, le syndrome b-thalassémique et l’hémoglobinose H. Le diagnostic moléculaire est un prérequis nécessaire à un éventuel diagnostic prénatal, qui permet aux couples qui le désirent de choisir de ne pas donner naissance à un enfant atteint de drépanocytose.
Le diagnostic utilise les mêmes techniques séparatives que le dépistage néonatal, à savoir l’électrophorèse et la chromatographie. Tout comme pour le dépistage, l’association de techniques différentes est nécessaire pour permettre l’identification des variants cliniquement significatifs : S, C, D-Punjab, E, O-Arab et Lepore. L’interprétation tient compte de l’évolution physiologique des différentes fractions d’hémoglobine au cours du développement. L’HbF majoritaire à la naissance va décroître et l’HbA va s’accroître pour atteindre des valeurs « adultes », respectivement HbF inférieure à 2 % et HbA égale à 95-98 % entre l’âge de 8 mois à 1 an. La fraction d’HbA2, fraction minoritaire, est difficilement interprétable chez l’enfant de moins de 6 mois ; elle atteint entre 8 mois et 1 an des valeurs « adultes » (HbA2 : 2,2-3,4 %). L’étude de l’hémoglobine des parents est importante pour préciser l’hémoglobinopathie de l’enfant. En effet, à partir de l’étude de l’hémoglobine, il est difficile chez l’enfant de moins de 1 an de faire le diag- nostic différentiel entre les formes S/S, S/b0-thalassémie ou encore S/PHHF (persistance héréditaire de l’hémoglobine F). Le diagnostic différentiel est essentiel car cette dernière association S-PHHF ne constitue pas un syndrome drépanocytaire majeur.
La confirmation diagnostique permet d’éliminer les rares faux positifs générés par le dépistage néonatal. Il s’agit essentiellement d’associations difficiles à dépister à la naissance : S-PHHF, S-Hope, S-K-Woolwich…
Les indications de diagnostic moléculaire sont limitées à la réalisation préalable d’une étude précise de l’hémoglobine. Les syndromes pathologiques sévères qui nécessitent une confirmation moléculaire sont le syndrome drépanocytaire majeur en l’absence d’étude familiale complète, le syndrome b-thalassémique et l’hémoglobinose H. Le diagnostic moléculaire est un prérequis nécessaire à un éventuel diagnostic prénatal, qui permet aux couples qui le désirent de choisir de ne pas donner naissance à un enfant atteint de drépanocytose.
Organisation de la prise en charge
Les enfants hétérozygotes A/S ne nécessitent pas de prise en charge médicale spécifique. La révélation du trait drépanocytaire peut cependant avoir des conséquences pour la famille. Les parents ont la possibilité de faire chacun une électrophorèse de l’hémoglobine afin de savoir s’ils ont un risque d’avoir un autre enfant atteint de syndrome drépanocytaire majeur. Cette révélation est anxiogène, et une information doit accompagner les familles qui le souhaitent. Compte tenu du grand nombre de familles concernées, la nécessité de disposer des ressources humaines suffisantes pour informer les parents reste une préoccupation.
Première consultation
La consultation doit se faire avec un médecin expert. Le rendez-vous est proposé par téléphone, sans que le mot « drépanocytose » ne soit prononcé car il est souvent vecteur d’une angoisse importante. Il ne sera énoncé qu’au cours de la consultation, lié à un discours rassurant sur la qualité de vie actuelle des enfants drépanocytaires.
Apprendre que son enfant a une drépanocytose est toujours un traumatisme majeur, même dans les cas assez rares où les parents se savaient porteurs du trait drépanocytaire. Il faut demander aux parents quelles représentations ils ont de la maladie (le mot drépanocytose n’est souvent pas connu par les parents, qui connaissent en revanche souvent l’expression « il est SS »). Pour la majorité des parents africains, être SS implique un décès dans l’enfance. Il faut donc présenter les progrès faits dans la prise en charge et inscrire l’enfant dans un projet de vie. Au mieux, cette annonce se fait en présence d’un psychologue.
L’organisation du parcours de soins est un des points majeurs de cette consultation. L’enfant doit être au cœur d’un réseau de soins maillant des acteurs de proximité et des équipes soignantes expertes. Les parents doivent être formés à devenir les premiers acteurs de soin, et notamment à reconnaître les signes exigeant une consultation dans un service hospitalier d’accueil des urgences : des symptômes potentiellement liés à la drépanocytose, une fièvre supérieure à 38,5 °C ; des signes évocateurs d’anémie sévère : pâleur brutale, refus de boire, somnolence ; la persistance de la douleur malgré la prise de paracétamol ; ainsi que les symptômes d’alerte pour tous les nourrissons, détresse respiratoire, diarrhée, vomissements…
On doit aussi vérifier que les parents sauront se rendre aux urgences, c’est-à-dire qu’ils savent appeler le Samu ou les pompiers, et ont prévu un mode de garde pour les autres enfants éventuels vivant au foyer. Des séances d’éducation thérapeutique ont été organisées dans de nombreux centres pédiatriques.
Les parents doivent au mieux avoir un médecin traitant, qu’il est utile de contacter pour lui rappeler les éléments principaux du suivi d’un enfant drépanocytaire. Des brochures d’information pour les médecins traitants sont disponibles auprès du réseau de soin ville-hôpital ROFSED.* Les services de protection maternelle et infantile sont aussi des relais essentiels pour le suivi préventif. Les médecins de crèche sont informés de la pathologie par un document de type « Protocole d’accueil individualisé ». Les médecins traitants peuvent participer à l’éducation thérapeutique et prendre en charge les douleurs modérées, les épisodes fébriles où la fièvre ne dépasse pas 38,5 °C chez les enfants âgés de moins de 3 ans, 39 °C chez ceux de plus de 3 ans.4 Ils peuvent contribuer à réfléchir avec les parents à leurs futurs choix reproductifs. Ils seront un pivot important au moment de la transition entre les services de soins pédiatriques et adultes. Le carnet de santé est un outil très utile où il est important de noter des faits marquants (génotype, taux d’hémoglobine à l’état basal, taille de la rate...) avec l’accord des parents.
Un entretien avec les services sociaux est souvent nécessaire. La drépanocytose est prise en charge par l’Assurance maladie à 100 %.
Apprendre que son enfant a une drépanocytose est toujours un traumatisme majeur, même dans les cas assez rares où les parents se savaient porteurs du trait drépanocytaire. Il faut demander aux parents quelles représentations ils ont de la maladie (le mot drépanocytose n’est souvent pas connu par les parents, qui connaissent en revanche souvent l’expression « il est SS »). Pour la majorité des parents africains, être SS implique un décès dans l’enfance. Il faut donc présenter les progrès faits dans la prise en charge et inscrire l’enfant dans un projet de vie. Au mieux, cette annonce se fait en présence d’un psychologue.
L’organisation du parcours de soins est un des points majeurs de cette consultation. L’enfant doit être au cœur d’un réseau de soins maillant des acteurs de proximité et des équipes soignantes expertes. Les parents doivent être formés à devenir les premiers acteurs de soin, et notamment à reconnaître les signes exigeant une consultation dans un service hospitalier d’accueil des urgences : des symptômes potentiellement liés à la drépanocytose, une fièvre supérieure à 38,5 °C ; des signes évocateurs d’anémie sévère : pâleur brutale, refus de boire, somnolence ; la persistance de la douleur malgré la prise de paracétamol ; ainsi que les symptômes d’alerte pour tous les nourrissons, détresse respiratoire, diarrhée, vomissements…
On doit aussi vérifier que les parents sauront se rendre aux urgences, c’est-à-dire qu’ils savent appeler le Samu ou les pompiers, et ont prévu un mode de garde pour les autres enfants éventuels vivant au foyer. Des séances d’éducation thérapeutique ont été organisées dans de nombreux centres pédiatriques.
Les parents doivent au mieux avoir un médecin traitant, qu’il est utile de contacter pour lui rappeler les éléments principaux du suivi d’un enfant drépanocytaire. Des brochures d’information pour les médecins traitants sont disponibles auprès du réseau de soin ville-hôpital ROFSED.* Les services de protection maternelle et infantile sont aussi des relais essentiels pour le suivi préventif. Les médecins de crèche sont informés de la pathologie par un document de type « Protocole d’accueil individualisé ». Les médecins traitants peuvent participer à l’éducation thérapeutique et prendre en charge les douleurs modérées, les épisodes fébriles où la fièvre ne dépasse pas 38,5 °C chez les enfants âgés de moins de 3 ans, 39 °C chez ceux de plus de 3 ans.4 Ils peuvent contribuer à réfléchir avec les parents à leurs futurs choix reproductifs. Ils seront un pivot important au moment de la transition entre les services de soins pédiatriques et adultes. Le carnet de santé est un outil très utile où il est important de noter des faits marquants (génotype, taux d’hémoglobine à l’état basal, taille de la rate...) avec l’accord des parents.
Un entretien avec les services sociaux est souvent nécessaire. La drépanocytose est prise en charge par l’Assurance maladie à 100 %.
Suivi ultérieur
Les nourrissons drépanocytaires de génotypes SS et Sb0thalassémiques doivent voir un médecin formé en drépanocytose environ tous les 3 mois dans leur première année de vie, ce rythme pouvant être moins fréquent, mais au moins annuel, chez les enfants SC et Sb+thalassémiques. Le rythme ultérieur de suivi dépend du génotype et de la sévérité de la maladie.
Des séances d’éducation thérapeutique, en groupe ou en individuel, doivent être systématiquement proposées. La connaissance du circuit d’urgence est contrôlée à chaque consultation.
La structuration des visites entre centres de proximité et centre de référence dépend beaucoup de la sévérité de la maladie, qui est parfois fluctuante au cours du temps chez le même enfant, et du lieu de résidence des parents. La plupart des familles habitent loin d’un centre de référence de la drépanocytose. Beaucoup de centres hospitaliers de proximité ont établi des réseaux avec les centres de référence, avec au mieux une circulation des dossiers informatisés entre eux. La répartition des soins doit être discutée pour chaque enfant. Les points les plus critiques sont souvent le lieu de réalisation des dopplers transcrâniens et des échanges transfusionnels en urgence. Il faut donc établir le parcours de soins pour chaque enfant et chaque « couple » centre de proximité-centre de référence, en incluant les médecins de ville.
Le Protocole national de soins des enfants drépanocytaires concourt à l’homogénéisation des prises en charge sur le territoire français.5
LABEL « MALADIE RARE »
Depuis 2004, la drépanocytose a été labellisée « maladie rare » par la Direction générale de l’offre de soins, qui a défini des centres de référence et des centres de compétence. La campagne de relabellisation en 2017 a redéfini deux centres de référence pour les « syndromes drépanocytaires majeurs, thalassémies et autres pathologies rares du globule rouge et de l’érythropoïèse » (l’un en région parisienne, l’autre aux Antilles), 13 sites constitutifs, 46 centres de compétence (arrêté du 8 août 2017 portant labellisation des réseaux des centres de référence prenant en charge les maladies rares). Ces structures, maillées aux laboratoires de diagnostic approfondi et aux associations de patients, sont regroupées dans la filière de santé maladies rares MCGRE (maladies constitutionnelles rares du globule rouge et de l’érythropoïèse), qui est présente sur l’ensemble du territoire national métropolitain et des départements et territoires d’outre-mer. V
Des séances d’éducation thérapeutique, en groupe ou en individuel, doivent être systématiquement proposées. La connaissance du circuit d’urgence est contrôlée à chaque consultation.
La structuration des visites entre centres de proximité et centre de référence dépend beaucoup de la sévérité de la maladie, qui est parfois fluctuante au cours du temps chez le même enfant, et du lieu de résidence des parents. La plupart des familles habitent loin d’un centre de référence de la drépanocytose. Beaucoup de centres hospitaliers de proximité ont établi des réseaux avec les centres de référence, avec au mieux une circulation des dossiers informatisés entre eux. La répartition des soins doit être discutée pour chaque enfant. Les points les plus critiques sont souvent le lieu de réalisation des dopplers transcrâniens et des échanges transfusionnels en urgence. Il faut donc établir le parcours de soins pour chaque enfant et chaque « couple » centre de proximité-centre de référence, en incluant les médecins de ville.
Le Protocole national de soins des enfants drépanocytaires concourt à l’homogénéisation des prises en charge sur le territoire français.5
LABEL « MALADIE RARE »
Depuis 2004, la drépanocytose a été labellisée « maladie rare » par la Direction générale de l’offre de soins, qui a défini des centres de référence et des centres de compétence. La campagne de relabellisation en 2017 a redéfini deux centres de référence pour les « syndromes drépanocytaires majeurs, thalassémies et autres pathologies rares du globule rouge et de l’érythropoïèse » (l’un en région parisienne, l’autre aux Antilles), 13 sites constitutifs, 46 centres de compétence (arrêté du 8 août 2017 portant labellisation des réseaux des centres de référence prenant en charge les maladies rares). Ces structures, maillées aux laboratoires de diagnostic approfondi et aux associations de patients, sont regroupées dans la filière de santé maladies rares MCGRE (maladies constitutionnelles rares du globule rouge et de l’érythropoïèse), qui est présente sur l’ensemble du territoire national métropolitain et des départements et territoires d’outre-mer. V
Références
1. Arrêté du 22 février 2018 relatif à l’organisation du programme national de dépistage néonatal recourant à des examens de biologie médicale. JORF n° 0049 du 28 février 2018, texte n° 18. www.legifrance.gouv.fr ou https://bit.ly/2HHtcJy
2. Bilan d’activité AFPDHE 2016. www.afdphe.org ou https://bit.ly/2UQuh5q
3. Haute Autorité de santé. Dépistage néonatal de la drépanocytose en France. Questions/Réponses. Service presse, mars 2014, www.has-sante.fr ou https://bit.ly/2TLYXsf
4. Haute Autorité de santé. Syndromes drépanocytaires majeurs de l’enfant et de l’adolescent. Protocole national de diagnostic et de soins pour une maladie rare. HAS, PNDS, janvier 2010.www.has-sante.fr ou https://bit.ly/2GX4dld
2. Bilan d’activité AFPDHE 2016. www.afdphe.org ou https://bit.ly/2UQuh5q
3. Haute Autorité de santé. Dépistage néonatal de la drépanocytose en France. Questions/Réponses. Service presse, mars 2014, www.has-sante.fr ou https://bit.ly/2TLYXsf
4. Haute Autorité de santé. Syndromes drépanocytaires majeurs de l’enfant et de l’adolescent. Protocole national de diagnostic et de soins pour une maladie rare. HAS, PNDS, janvier 2010.www.has-sante.fr ou https://bit.ly/2GX4dld
Dans cet article

Summary Newborn screening of sickle cell disease and management of care
Le dépistage néonatal de la drépanocytose est systématique chez tous les nouveau-nés des départements, régions et collectivités d’outre-mer français, et il est ciblé en métropole chez les nouveau-nés dont les parents appartiennent à un groupe à risque pour la drépanocytose. Le taux de ciblage va de 9,1 % en Bretagne à 73,6 % en Île-de-France. Le dépistage néonatal est réalisé à partir d’un échantillon de sang total obtenu par piqûre au talon du nouveau-né et déposé sur un buvard. L’analyse est fondée sur des techniques séparatives, électrophorèse et chromatographie, permettant d’une part de séparer les hémoglobines en fonction de leurs caractéristiques physicochimiques et d’autre part de quantifier les différentes fractions d’hémoglobine. Tout résultat positif doit être systématiquement vérifié à l’aide d’une technique différente de celle utilisée en première intention. Les résultats des nouveau-nés hétérozygotes, qui ne sont pas malades, sont rendus aux parents par simple courrier ou au décours d’une consultation selon les organisations locales. Les résultats des nouveau-nés possiblement atteints sont adressés au pédiatre référent. Celui-ci organise une consultation d’annonce avec les parents et le nouveau-né. La première consultation doit être faite par un médecin expert. Tout enfant doit être au cœur d’un réseau de soins maillant des acteurs de proximité et des équipes soignantes expertes. Les parents doivent être formés à devenir les premiers acteurs de soin, et notamment à reconnaître les signes exigeant une consultation dans un service hospitalier d’accueil des urgences. En 2016, 431 enfants ont été dépistés, soit une incidence de la maladie à la naissance de 1/771 dans la population testée.