Reconnaître une dystonie
La dystonie peut concerner presque toutes les régions du corps, apparaître à n’importe quel âge, classiquement de façon progressive mais parfois brutalement, et s’accompagner d’autres manifestations neurologiques (myoclonies, spasticité, syndrome cérébelleux ou extrapyramidal…).
Les atteintes généralisées sont plus fréquentes chez l’enfant, contrairement aux formes focales ou segmentaires qui touchent davantage l’adulte (crampe de l’écrivain, torticolis spasmodique, blépharospasme).
Chez le nourrisson, on peut constater initialement une hypotonie axiale, suivie d’une dystonie périphérique. Les formes secondaires affectent plus volontiers l’enfant, ce qui incite à une recherche poussée de l’étiologie (IRM notamment). Si la dystonie se manifeste à cet âge, elle tend à s’aggraver avec le temps. En revanche, celle apparaissant chez l’adulte, plutôt focale ou segmentaire, reste limitée à la partie du corps touchée.
Pour faciliter le diagnostic, une classification récente a été établie selon 2 grands axes, le premier est clinique, le second étiologique (tableau).1 Certaines dystonies sont acquises, d’autres sont génétiques ou hérédodégénératives, mais le plus souvent, elles restent idiopathiques.
La prise de drogues et toxiques est à rechercher à l’interrogatoire (neuroleptiques, lithium, cocaïne…).
Une IRM est nécessaire pour vérifier l’absence de lésion cérébrale, notamment au niveau des noyaux gris centraux, ou chercher des éléments d’orientation (anoxie cérébrale, pathologie métabolique…).
Dans les formes focales, idiopathiques, typiques, l’IRM n’est pas systématique.
Chez l’adulte,les plus fréquentes sont dites primaires (dystonie isolée cliniquement avec un bilan complémentaire négatif). Deux groupes principaux : les dystonies focales, peu évolutives, et les primaires liées à un gène identifié comme la dystonie DYT1 survenant avant 26 ans et représentant plus de 60 % des formes débutant au niveau d’un membre.
Dystonies focales
Premières manifestations : des difficultés à réaliser facilement un mouvement (rotation de la tête, écriture) avec une impression de tension musculaire, rarement douloureuse.
Photosensibilité, intolérance au vent avec larmoiement, puis clignements involontaires peuvent révéler un blépharospasme (qui affecte les muscles des paupières).
Les symptômes sont la plupart du temps continus excepté dans les dystonies de fonction comme la crampe de l’écrivain (qui survient en écrivant, parfois dès la saisie du stylo ou après un délai) ou la dystonie du musicien qui ne se manifeste que lorsque le patient joue de son instrument.
Ils peuvent disparaître spontanément pendant quelque temps (rémission) et parfois à plusieurs reprises, mais ce phénomène est transitoire.
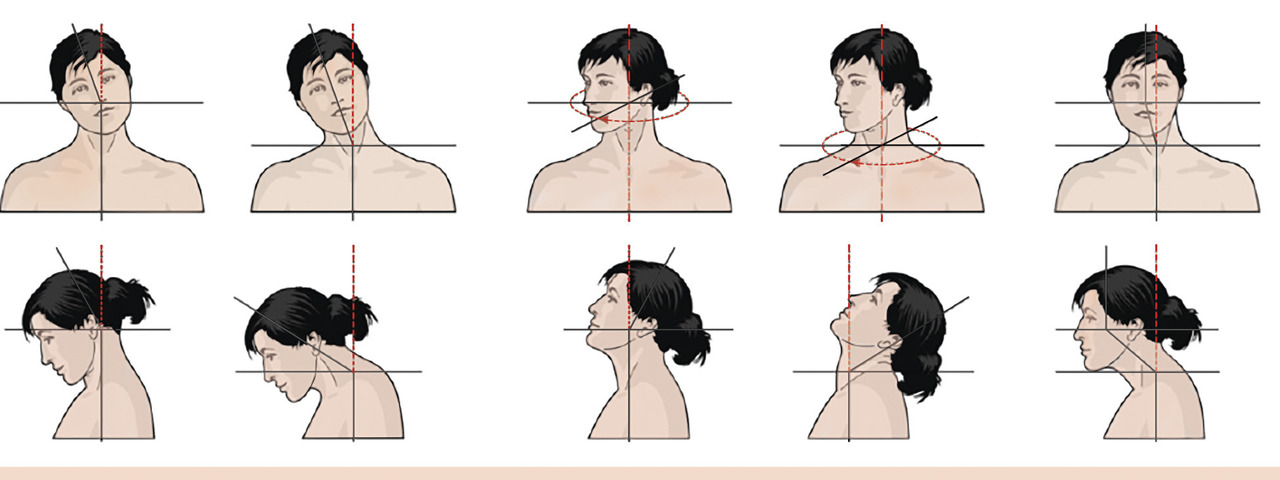
Dystonie cervicale
Blépharospasme
Le symptôme survient préférentiellement lorsque le sujet (classiquement âgé de plus de 50 ans) est actif (conduite automobile, marche à l’extérieur), dans un milieu lumineux. Il est soulagé par le repos et l’obscurité. D’autres muscles du visage peuvent aussi être atteints (ceux de la dystonie oromandibulaire), donnant le syndrome de Meige.
Dystonie de fonction
Dysphonie spasmodique
Dystonie oromandibulaire
Affections métaboliques et hérédodégénératives
Ces dernières années, un grand nombre de gènes associés ont été identifiés. Les diagnostics, très nombreux et aux phénotypes variables, ne sont pas abordés ici.
Parmi les causes, de plus en plus de pathologies curables sont identifiées (déficit intracérébral en folates, en créatinine, en transporteur du glucose, maladie de Niemann-Pick type C…). La dystonie sensible à la L-dopa et la maladie de Wilson sont à connaître.
Dystonie sensible à la L-dopa
– des troubles de la marche dans l’enfance avec une dystonie des membres inférieurs s’aggravant en fin de journée ;
– toute dystonie, surtout en cas de fluctuations et de signes parkinsoniens, avec une IRM cérébrale normale ;
– une encéphalopathie précoce avec signes dysautonomiques.
Le tableau clinique peut être similaire à celui de la paralysie cérébrale infantile, ou à celui d’une dystonie focale débutant à l’âge adulte, ou même à un syndrome parkinsonien de l’adulte. La prévalence en Europe est estimée à 1-5 par million d’habitants, mais il faut y penser car elle est traitable facilement !
En cas de suspicion, il faut faire un test thérapeutique à la L-dopa : on débute avec de petites doses : 0,5 à 1 mg/kg et on augmente progressivement jusqu’à un pallier de 5 mg/kg pendant 4 semaines, puis si besoin, jusqu’à 600 mg chez l’adulte. L’essai doit durer au moins 3 mois car si certains ressentent rapidement l’effet de la dopa, d’autres l’éprouveront plus tard.
Le patient doit être adressé dans un centre spécialisé pour ponction lombaire avec étude des neurotransmetteurs du LCR.
Maladie de Wilson
Les formes neurologiques s’observent volontiers chez l’adolescent plus âgé. Le tableau est alors très polymorphe, associant à des degrés variables tremblement, dysarthrie, dystonie focale notamment faciale – donnant un sourire sardonique – ou généralisée, troubles de l’écriture, de la déglutition. Les affections psychiatriques sont fréquentes, syndrome dépressif en particulier.
Prise en charge
Une démarche centrée sur le bien-être, la gestion du stress (sophrologie, yoga, prise en charge psychologique) est capitale en raison de l’impact de la maladie sur la qualité de vie, le stress étant un facteur aggravant.
Pour les dystonies focales, la toxine botulique, traitement de référence administré tous les 3 mois, est très efficace, en particulier dans le torticolis spasmodique et le blépharospasme. Injectée dans les muscles cervicaux, elle peut provoquer des troubles de la déglutition ; dans l’orbiculaire des paupières : ptosis possible.
Dans les formes sévères, les dystonies généralisée, ou focales, sous certaines conditions, une stimulation cérébrale profonde peut être proposée.
La chirurgie a des indications très limitées (dénervation périphérique dans la dystonie cervicale…).
Une prise en charge en service de neurologie spécialisé dans les pathologies du mouvement est recommandée.
Formes particulières à connaître
Certaines dystonies se manifestent de façon paroxystique, le plus souvent chez le sujet jeune. La sémiologie et le caractère fluctuant des troubles ne doivent pas faire oublier les causes organiques ou iatrogènes sous-jacentes :
• les dystonies paroxystiques durant quelques secondes, minutes ou heures (principalement génétiques, occasionnellement symptomatiques) ;4
• les médicaments (Primpéran) ;
• les dyskinésies paroxystiques kinésigéniques ou non kinési- géniques d’origine génétique, induites par l’exercice ;4
• la SEP ou un AVC ;
• les mitochondriopathies (dont le déficit en pyruvate déshydro- génase) ;
• l’hypoparathyroïdie, l’hypothyroïdie, le trouble du transfert de la T3 ;
• l’hémiplégie alternante liée au gène ATP1A3.
La dystonie DYT12 liée également à une mutation sur le gène ATP1A3 peut s’installer rapidement en quelques heures, au décours d’un stress, et être prise à tort pour une manifestation neurofonctionnelle.4
2. Macerollo A, Superbo M, Gigante AF, Livrea P, Defazio G. Diagnostic delay in adult-onset dystonia: data from an Italian movement disorder center. J Clin Neurosci 2015;22:608-10.
3. Müller J, Wissel J, Masuhr F, Ebersbach G, Wenning GK, Poewe W. Clinical characteristics of the geste antagoniste in cervical dystonia. J Neurol 2001;248:478-82.
4. Méneret A, Roze E. Paroxysmal movement disorders: An update. Rev Neurol (Paris) 2016;172:433-45.
Dans cet article
 Encadrés
Encadrés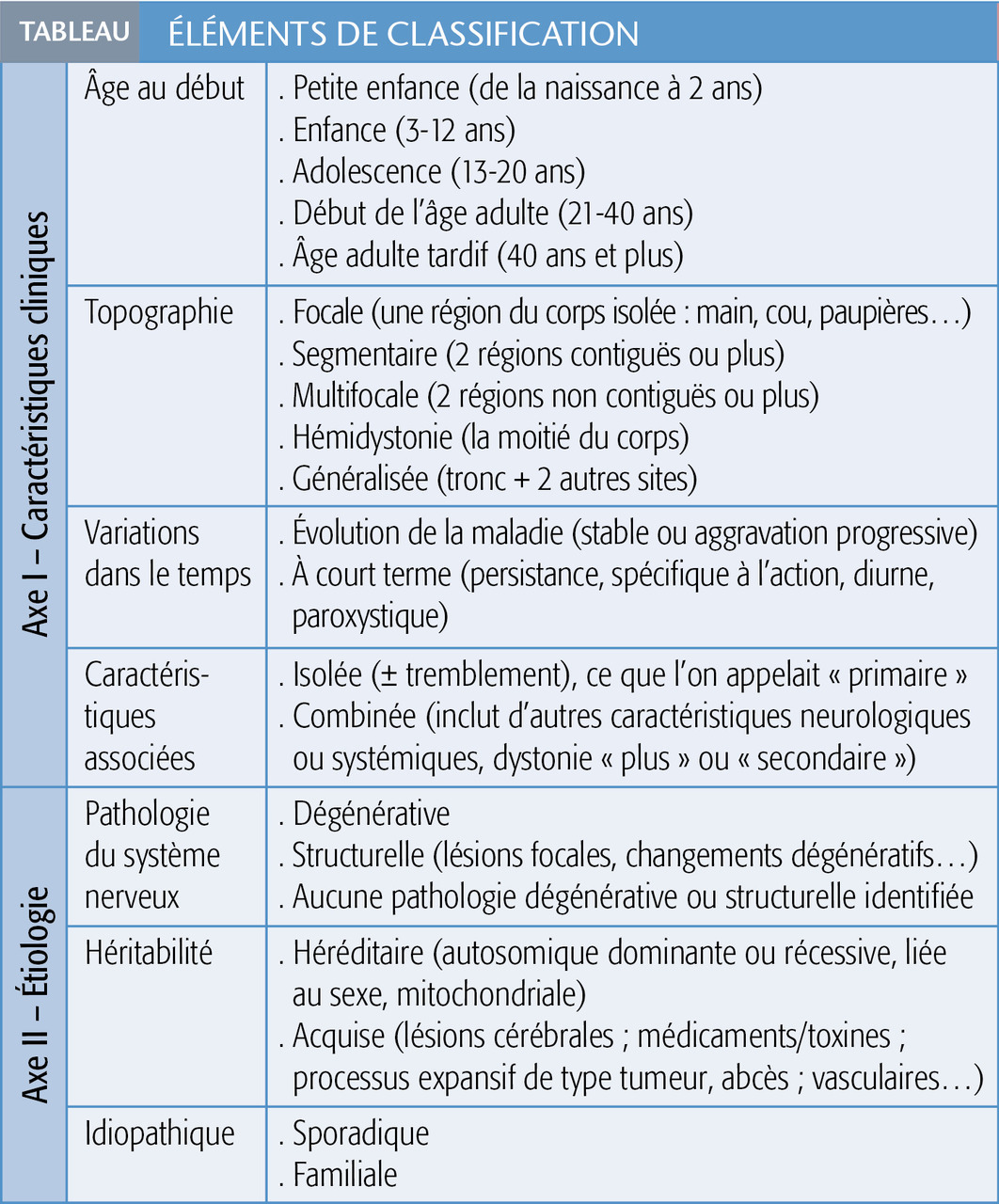

Avant 3 ans : dystonie rarement isolée, chercher une cause hérédodégénérative +++.
Entre 3 et 26 ans : parfois isolée, elle tend à se généraliser et peut avoir une explication génétique (dystonie primaire de type DYT1). Chercher aussi une cause neurologique.
Après 26 ans, plus souvent focale ou segmentaire, elle peut être primaire ou secondaire. En présence de tableaux neurologiques plus complexes, évoquer une cause hérédodégénérative.
IRM si sujet jeune, distribution hémicorporelle, autres signes neurologiques, installation rapide (en l’absence de prise de neuroleptique).
Adresser à un neurologue spécialisé dans les troubles du mouvement.
Prise en charge multidisciplinaire (kiné, psychologue, neurologue) à 100 % (ALD hors liste).