Connaitre les principales hypothèses diagnostiques devant une hyperéosinophilie et les premiers examens complémentaires les plus pertinents.
Savoir identifier un syndrome d'hypersensibilité médicamenteuse sévère.
Introduction
L’augmentation des éosinophiles peut s’intégrer principalement dans 4 grands sous-groupes étiologiques : l’allergie, les causes iatrogènes, les causes parasitaires et certaines néoplasies solides ou hématologiques. Plus rarement, elle peut être le signe de certaines maladies immunologiques, de certains déficits immunitaires, d’autres causes diverses, ou entrer dans le cadre des hyperéosinophilies primaires, des éosinophilies de signification incertaine et des syndromes hyperéosinophiliques idiopathiques en cas d’atteinte d’organe associée.
Par définition, l’éosinophilie correspond à une augmentation du chiffre des polynucléaires éosinophiles sanguins à plus de 500/mm3, et l’hyperéosinophilie à un taux supérieur à 1 500/mm3.
Le polynucléaire éosinophile
La maturation médullaire de cette cellule myéloïde est contrôlée par divers facteurs de transcription (notamment GATA-1, PU.1 et c/EBP), et par les « éosinopoïétines » (GM-CSF, interleukine-3 [IL-3] et IL-5). L’IL-5 est la cytokine la plus spécifique de la lignée éosinophile ; sa production est assurée par cette même cellule, par les mastocytes, par les basophiles et par les lymphocytes Th2.
Après une stimulation antigénique (micro-organismes, allergènes, médiateurs de l’inflammation, fragment Fc des IgA/E/G, complexes immuns ou cytokines), le polynucléaire éosinophile peut exercer différentes actions :
- cytotoxique directe sur les parasites ou les cellules cancéreuses ;
- pro-inflammatoire, avec recrutement d’autres cellules inflammatoires par le biais de la sécrétion de diverses cytokines Th1 ou Th2 ;
- immunorégulatrice adaptative par le biais d’une production de cytokines (Th2) ou de modulations locales de l’activité lymphocytaire après une présentation antigénique.
Il n’y a pas de correspondance stricte entre l’éosinophilie sanguine et l’importance de l’infiltration éosinophilique tissulaire, qu’elle soit locale au sein d’un foyer inflammatoire spécifique ou systémique. L’infiltration systémique éosinophile est possible au niveau de tout organe, mais préférentiellement dans les tissus pulmonaire, myocardique et cutané.
Deux mécanismes pathogéniques peuvent concourir à la survenue d’une éosinophilie :
- un processus réactionnel inflammatoire local (invasion parasitaire) ou systémique (allergie médicamenteuse), médié par une augmentation non spécifique des diverses cytokines régulatrices de la production médullaire de l’éosinophile ;
- un processus prolifératif clonal soit de l’éosinophile lui-même, soit de cellules productrices de la cytokine majeure de sa régulation (en l’occurrence, le lymphocyte T sécrétant de l’IL-5).
Classification étiologique des éosinophilies
Éosinophilies réactionnelles (ou secondaires)
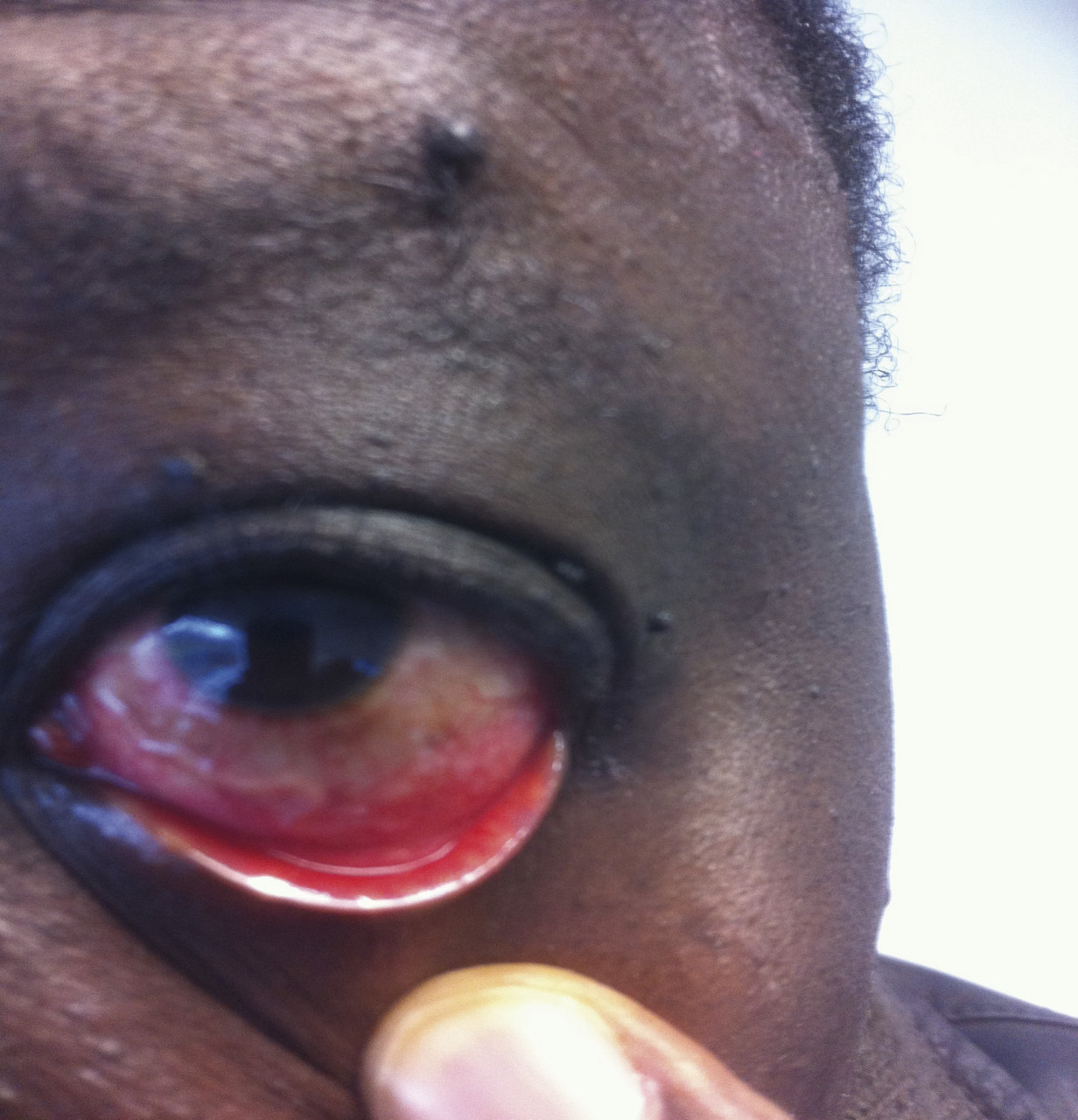
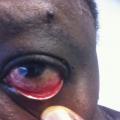
La circonstance de découverte est, le plus souvent, la mise en évidence fortuite d’une éosinophilie modérée. Plus rarement, il s’agit d’une hyperéosinophilie (> 1 500/mm3), pouvant s’accompagner de symptômes. Ceux-ci peuvent être liés à la maladie sous-jacente ou à l’infiltration tissulaire avec, éventuellement, une dysfonction d’organe. L’exemple le plus illustratif est l’éosinophilie périodique correspondant aux phases de migration tissulaire d’une parasitose digestive, d’une filariose (
Dans le cadre des syndromes hyperéosinophiliques en rapport avec une hyperactivation lymphocytaire T (variant lymphoïde du syndrome hyperéosinophilique), il s’agit d’une population clonale ou activée de lymphocytes T, le plus souvent CD4, qui sécrète des quantités excessives d’IL-5 responsables d’une prolifération anormale des éosinophiles. Une hyper-IgE et des manifestations atopiques et/ou des angio-œdèmes (syndrome de Gleich) sont caractéristiques de cette entité. Les récentes techniques de biologie moléculaire et d’immunophénotypage permettent la détection le plus souvent d’un clone ou d’une population activée de lymphocytes T CD4 positifs, en règle minime, ne représentant que de 1 à 4 % des lymphocytes T, qui se caractérisent par un trou phénotypique sur l’expression du CD3 (lymphocytes T CD3– et CD4+). Plus rarement, il peut s’agir de lymphocytes T CD3+ CD4– et CD8– ou CD3+ CD7–.
Éosinophilies primaires (clonales)
Il peut aussi s’agir d’authentiques syndromes myéloprolifératifs s’accompagnant d’une éosinophilie, comme une leucémie myélomonocytaire chronique ou une leucémie myéloïde aiguë ou chronique.
Éosinophilie idiopathique
Physiopathologie et manifestations cliniques communes
Les atteintes les plus précoces et les plus fréquentes sont bronchopulmonaires (pleurésies éosinophiliques, infiltrats interstitiels…). Les atteintes les plus graves sont cardiaques (insuffisance cardiaque par fibrose endomyocardique, myocardite aiguë, thromboses, valvulopathies…) et neurologiques (encéphalopathie, atteintes focales…). Des atteintes cutanées (prurit, maculo-papules…), digestives (malabsorption, hépatomégalie, pancréatite, ascite éosinophilique), rénales, musculaires ou rétiniennes sont également décrites. Des complications thromboemboliques peuvent aussi survenir. Le pronostic vital peut être engagé en moins de six mois, essentiellement par les atteintes respiratoires (détresse respiratoire aiguë), cardiaques (fibrose endomyocardique) et neurologiques. Parfois, une atteinte viscérale ne survient qu’après plusieurs années d’évolution.
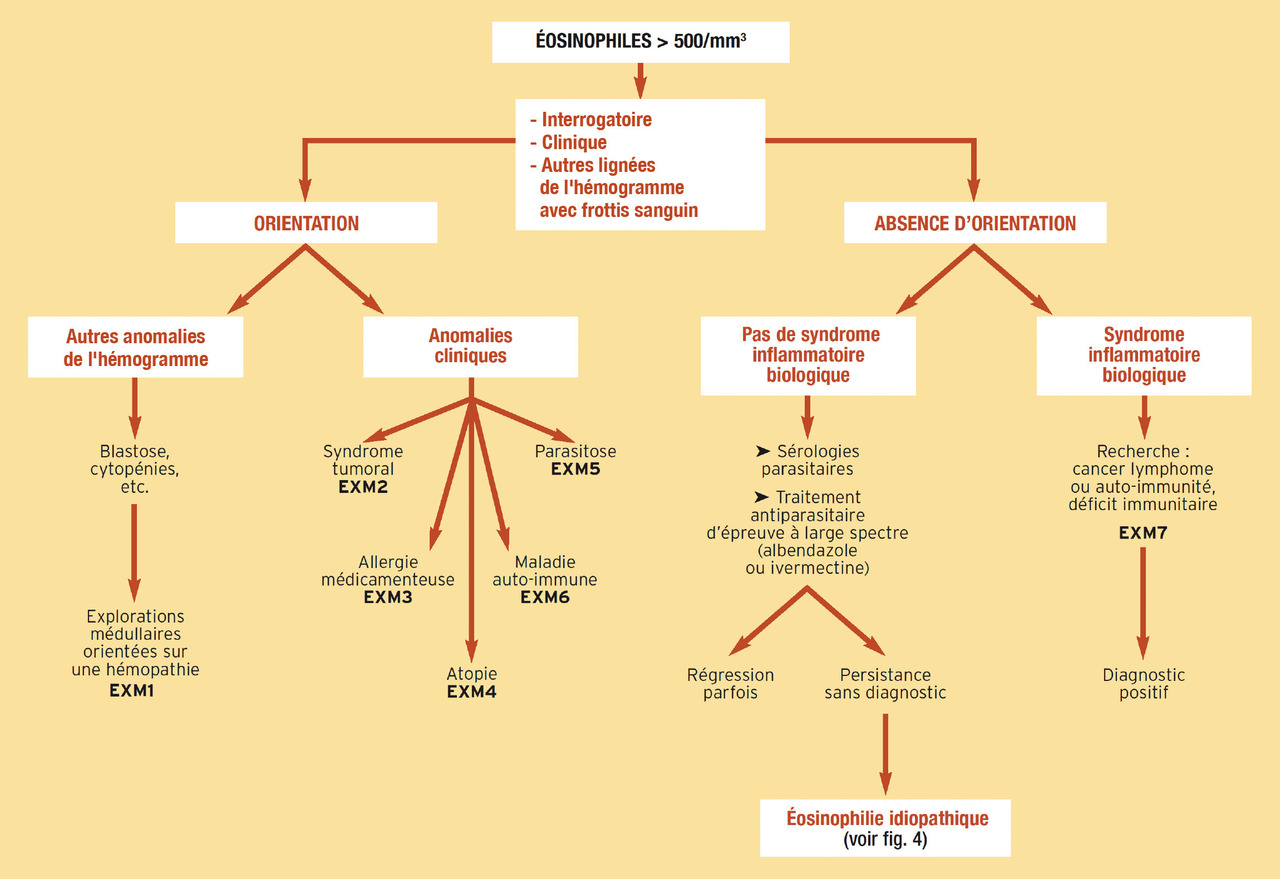
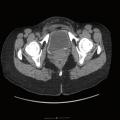
Conduite diagnostique pratique et principes du traitement (fig. 3 et 4)
Dans le même temps, les diagnostics différentiels de syndrome myéloprolifératif aigu de type leucémie à éosinophiles sont écartés, et la recherche d’éventuelles atteintes organiques (cœur, poumons, foie…) est entreprise.
Un traitement d’épreuve antiparasitaire large (ivermectine ou albendazole), un scanner thoraco-abdomino-pelvien et/ou une endoscopie digestive, à la recherche d’un cancer profond, sont discutés à la fin de cette première étape diagnostique, en l’absence d’autre point d’appel clinique.
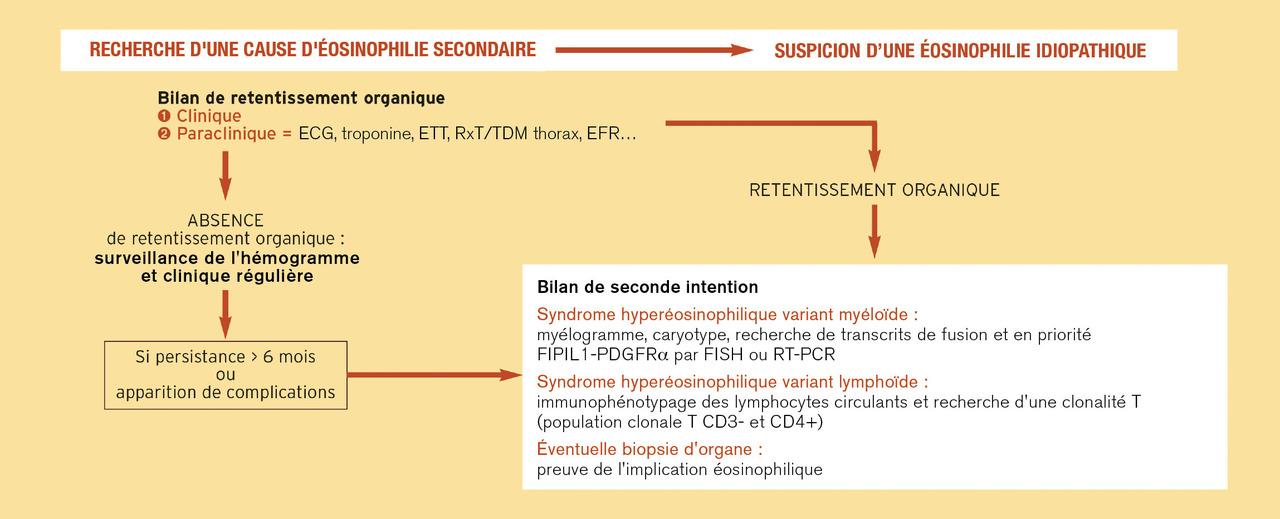
En cas de négativité de cette première démarche et de persistance de l’éosinophilie à des valeurs supérieures à 1 500/mm3 pendant plus de six mois, des examens spécialisés clôturent cette étape investigatrice : biopsie ostéomédullaire, myélogramme et caryotype médullaire ; biologie moléculaire et techniques d’immunophénotypage lymphocytaire.
En fonction de ce bilan, la conduite à tenir est adaptée.
En l’absence de diagnostic étiologique et de retentissement clinique, le diagnostic, parfois provisoire, d’éosinophilie de signification incertaine est retenu, et une simple surveillance suffit, avec évaluation systématique tous les six mois d’un éventuel retentissement organique.
En l’absence de diagnostic étiologique mais en présence d’un retentissement fonctionnel général (fièvre, altération de l’état général) ou organique, le diagnostic de syndrome hyperéosinophilique idiopathique est posé, et une corticothérapie en première ligne est licite.
En cas d’identification d’une cause d'éosinophilie secondaire, le pronostic des formes est souvent bon et lié à l’efficacité du traitement de cette cause. En dehors des causes parasitaires, une corticothérapie d’appoint peut être associée au traitement de la maladie causale.
En cas d’identification d’un syndrome hyperéosinophilique de type myéloïde ou lymphoïde, le pronostic est très difficile à établir ; il varie en effet selon les formes pathogéniques du syndrome hyperéosinophilique, l’importance de l’infiltration tissulaire (qui n’est pas toujours corrélée à celle de l’éosinophilie sanguine), la possibilité ou non d’un traitement ciblé et selon les atteintes d’organes.
Le traitement du syndrome hyperéosinophilique de type myéloprolifératif dépend de l’anomalie moléculaire identifiée (imatinib si transcrit FIP1L1-PDGFRα, FIP1L1-PDGFRβ ou ETV6-ABL1 ; sunitinib ou sorafénib si transcrit ETV6-FLT3…) et peut comprendre, en dernier recours, l’allogreffe de cellules souches hématopoïétiques.
Dans le syndrome hyperéosinophilique lymphoïde, une régression de l’éosinophilie est souvent observée en une semaine sous cortisone. Le pronostic est fréquemment meilleur, hormis la possible évolution de l’anomalie de fond vers un syndrome lymphoprolifératif. En cas d’échec des corticoïdes ou de corticodépendance, les immunosuppresseurs (interféron, ciclosporine…), les traitements myélosuppressifs comme l’hydroxycarbamide ou les anticorps monoclonaux anti-IL-5 (dont le mépolizumab) peuvent être utilisés.
Le syndrome d’hypersensibilité médicamenteuse sévère
Il peut se manifester par une phase prodromique (hyperthermie, adénopathies, syndrome pseudogrippal, prurit) jusqu’à deux semaines avant l’apparition de l’atteinte cutanée. Celle-ci peut associer un œdème facial ou distal, une érythrodermie, un purpura, des pustules et parfois une atteinte focale des muqueuses. La gravité de ce syndrome tient à la présence d’atteintes d’organes (hépatite, néphropathie interstitielle, pneumopathie interstitielle, myocardite, syndrome d’activation lymphohistiocytaire, notamment). Outre l’éosinophilie, le bilan biologique peut mettre en évidence des lymphocytes activés, une cytolyse hépatique, voire une cholestase, une insuffisance rénale, une réactivation des virus de la famille des Herpèsvirus (HHV6, HHV7, EBV, CMV) ou du parvovirus B19. Les résultats anatomopathologiques d’une biopsie cutanée, indispensable pour éliminer les diagnostics différentiels, sont variables et non pathognomoniques dans le DRESS syndrome.
Le traitement, non codifié en dehors de l’arrêt du médicament incriminé, fait principalement appel aux corticoïdes locaux et/ou systémiques en fonction de la gravité du tableau clinicobiologique. On y associe un traitement symptomatique (antipyrétiques, émollients, antiprurigineux…).
La mortalité est estimée entre 5 et 10 %, principalement liée aux complications cardiaques, pulmonaires ou à un syndrome d’activation lymphohistiocytaire. La régression de l’atteinte cutanée est lente, sur plusieurs semaines, avec des poussées cutanées et/ou viscérales possibles au cours de l’évolution, même en l’absence de réintroduction du médicament incriminé.
POINTS FORTS À RETENIR
Dans l’exploration étiologique d’une éosinophilie, l’interrogatoire, étape essentielle de l’examen clinique, cherche à faire le point sur des voyages éventuels en pays tropicaux, quelle que soit leur ancienneté. Le cas échéant, la strongyloïdose (seule nématodose ayant un cycle interne d’autoréinfestation avec pérennisation possible sur de nombreuses décennies et possible dissémination multiviscérale mortelle chez l’immunodéprimé) doit être recherchée en priorité (parasitologie des selles sur 3 jours avec enrichissement) et systématiquement traitée (ivermectine) avant toute thérapeutique immunosuppressive.
Dans la démarche diagnostique, les causes d'éosinophilies secondaires, de loin les plus fréquentes, doivent être systématiquement passées en revue. Le fil conducteur peut en être l’existence ou non d’antécédents (atopie ?), d’autres anomalies de l’hémogramme (myélémie ou basophilie orientant vers une leucémie myéloïde aiguë ou chronique), d’un syndrome inflammatoire biologique (cancers, maladies auto-immunes ou parasitose, comme la trichinose avec augmentation des enzymes musculaires…). Le caractère fluctuant de l’éosinophilie oriente vers des parasitoses digestives avec un cycle de migration tissulaire.
Dans un second temps, si l’éosinophilie persiste plus de six mois avec des valeurs très élevées (plus de 1 500/mm3), les syndromes hyperéosinophiliques de type myéloïde (augmentation de la vitamine B12, de l’acide urique, splénomégalie, biopsie ostéomédullaire, myélogramme, caryotype, transcrit FIP1L1-PDGFRα…) ou lymphoïde (augmentation importante des IgE, recherche d’un clone T par immunophénotypage lymphocytaire et biologie moléculaire) sont à rechercher.
Dans de nombreux cas, aucune cause n’est trouvée, et la persistance d’une hyperéosinophilie après traitement d’épreuve antiparasitaire large (ivermectine et albendazole notamment) fait retenir une probable forme idiopathique. Une surveillance clinique et complémentaire veille à repérer tout retentissement viscéral qui nécessiterait un traitement.
Klion AD. How I treat hypereosinophilic syndromes. Blood 2015;126: 1069-77.
Valent P, Degenfeld-Schonburg L, Sadovnik I, et al. Eosinophils and eosinophil-associated disorders: immunological, clinical, and molecular complexity. Semin Immunopathol 2021;43:423-38.
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. 2016;127:2391-405.
Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012;130:607-12.
Duong TA, Valeyrie-Allanore L, Wolkenstein P, et al. Severe cutaneous adverse reactions to drugs. Lancet 2017;390:1996-2011.
CEDEF. Item 322 – UE 10 Iatrogénie. Diagnostic et prévention : toxidermies médicamenteuses. Ann Dermatol Venereol 2018;145 Suppl 1:S189-S200.
Cabañas R, Ramírez E, Sendagorta E, et al. Spanish Guidelines for Diagnosis, Management, Treatment, and Prevention of DRESS Syndrome. J Investig Allergol Clin Immunol 2020;30:229-53.
Vous pouvez retrouver un quiz lié à cet item sur notre site internet : https://www.larevuedupraticien.fr/les-tests/les-quiz
 Encadrés
Encadrés