Objectifs
Diagnostiquer les principales formes d’épilepsie de l’enfant et de l’adulte.
Identifier les situations d’urgence.
Connaître les principes de la prise en charge.
Identifier les situations d’urgence.
Connaître les principes de la prise en charge.
L’épilepsie est une affection caractérisée par la répétition chronique de crises épileptiques, expression clinique de décharges neuronales excessives. Cette prédisposition durable à générer des crises d’épilepsie entraîne des conséquences neurobiologiques, cognitives, psychologiques et sociales. On retrouve deux pics d’incidence de l’épilepsie dans la vie : avant l’âge de 10 ans (autour de 70/100 000) et au-delà de 80 ans (autour de 80/100 000). Il s’agit de la première maladie neurologique de l’enfant.
Il est important de distinguer :
La prise en charge thérapeutique varie en conséquence, s’attache également au suivi des comorbidités et à favoriser l’insertion sociale de l’enfant.
Il est important de distinguer :
- les crises provoquées, en réaction à une situation telle qu’une hypoglycémie, une infection du système nerveux, un trouble ionique, une syncope cardiaque, une anoxie, un traumatisme crânien… Les crises provoquées, notamment fébriles, sont plus fréquentes chez l’enfant que chez l’adulte ;
- les crises d’épilepsie survenant de façon spontanée ou parfois lors de dette de sommeil.
La prise en charge thérapeutique varie en conséquence, s’attache également au suivi des comorbidités et à favoriser l’insertion sociale de l’enfant.
Poser le diagnostic
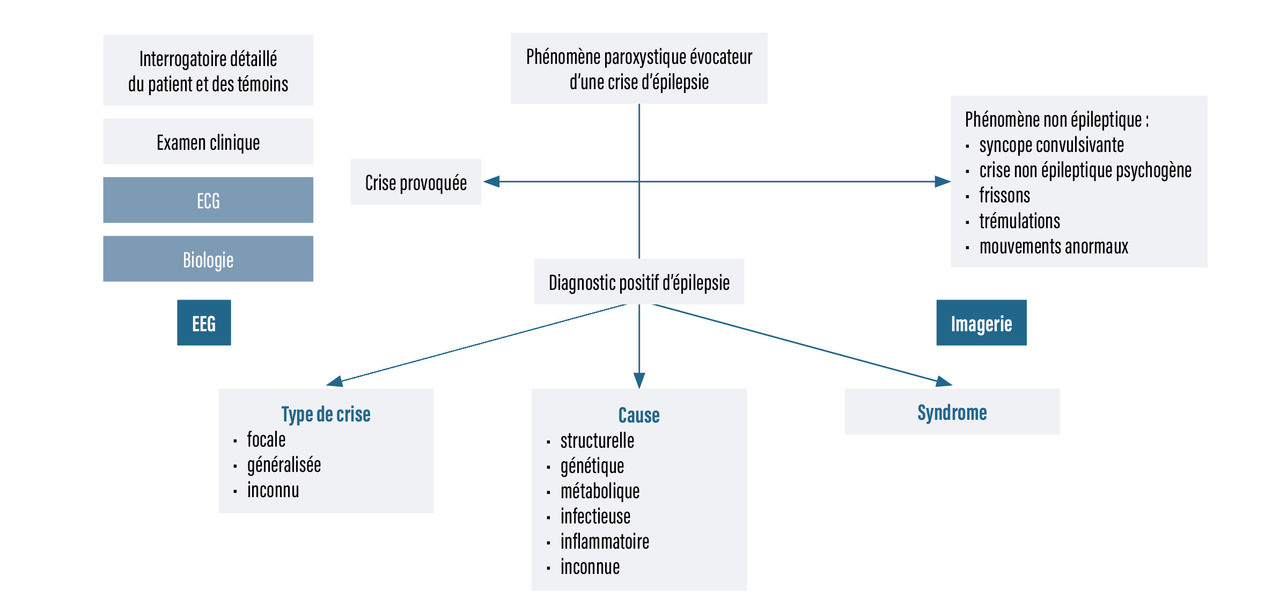
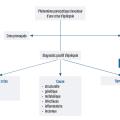
Le diagnostic d’une épilepsie se fait en deux étapes (fig. 1 ) :
- diagnostic positif de crise d’épilepsie, avec élimination des diagnostics différentiels ;
- diagnostic positif de maladie épileptique, syndromique et étiologique.
Diagnostic positif de crise d’épilepsie
Il est rare d’assister à une crise d’épilepsie, et l’interrogatoire de l’entourage et de l’enfant ou la relecture de vidéos familiales ont une place essentielle. Il est nécessaire de recueillir les circonstances de survenue, un éventuel signal-symptôme, les signes de la phase d’état, puis post-critiques : symptômes sensitivo-moteurs, localisation, état de conscience, symptômes dysautonomiques, émotionnels, cognitifs, sensoriels, et leur durée.
Aucun signe clinique n’est pathognomonique de crise d’épilepsie, même si certains sont hautement évocateurs (par exemple : crise tonico-clonique, ou spasmes en salves chez le nourrisson). Dans le cas d’une crise sans perte de connaissance, c’est souvent le caractère répété, stéréotypé, transitoire, non déclenché des symptômes qui permet d’évoquer le diagnostic.
Aucun examen biologique n’est recommandé pour le diagnostic positif de crise d’épilepsie. Dans certains cas, un électroencéphalogramme (EEG) vidéo de longue durée est nécessaire pour affirmer le diagnostic.
Il faut distinguer les crises focales qui impliquent une région cérébrale et les crises généralisées qui impliquent l’ensemble du cortex. Une crise focale peut évoluer vers une crise généralisée, décrite alors comme secondairement généralisée. Certaines crises sont inclassables.
La sémiologie des crises épileptiques focales varie en fonction de la topographie de la zone épileptogène et de l’âge de l’enfant : symptômes sensitifs ou moteurs focaux, symptômes sensoriels subjectifs avec hallucinations, automatismes oromoteurs, gestuels, symptômes émotionnels, dysautonomiques. Certaines crises, notamment frontales, peuvent n’apparaître que durant le sommeil.
Enfin, il existe de nombreux phénomènes moteurs ou comportementaux paroxystiques non épileptiques : mouvements anormaux paroxystiques, syncopes, migraines accompagnées, crises non épileptiques psychogènes, trémulations, myoclonies du sommeil, spasmes du sanglot, frissons… Ces manifestations doivent être connues afin d’éviter un diagnostic d’épilepsie par excès.
Aucun signe clinique n’est pathognomonique de crise d’épilepsie, même si certains sont hautement évocateurs (par exemple : crise tonico-clonique, ou spasmes en salves chez le nourrisson). Dans le cas d’une crise sans perte de connaissance, c’est souvent le caractère répété, stéréotypé, transitoire, non déclenché des symptômes qui permet d’évoquer le diagnostic.
Aucun examen biologique n’est recommandé pour le diagnostic positif de crise d’épilepsie. Dans certains cas, un électroencéphalogramme (EEG) vidéo de longue durée est nécessaire pour affirmer le diagnostic.
Il faut distinguer les crises focales qui impliquent une région cérébrale et les crises généralisées qui impliquent l’ensemble du cortex. Une crise focale peut évoluer vers une crise généralisée, décrite alors comme secondairement généralisée. Certaines crises sont inclassables.
La sémiologie des crises épileptiques focales varie en fonction de la topographie de la zone épileptogène et de l’âge de l’enfant : symptômes sensitifs ou moteurs focaux, symptômes sensoriels subjectifs avec hallucinations, automatismes oromoteurs, gestuels, symptômes émotionnels, dysautonomiques. Certaines crises, notamment frontales, peuvent n’apparaître que durant le sommeil.
Enfin, il existe de nombreux phénomènes moteurs ou comportementaux paroxystiques non épileptiques : mouvements anormaux paroxystiques, syncopes, migraines accompagnées, crises non épileptiques psychogènes, trémulations, myoclonies du sommeil, spasmes du sanglot, frissons… Ces manifestations doivent être connues afin d’éviter un diagnostic d’épilepsie par excès.
Diagnostic positif de maladie épileptique
Une maladie épileptique peut être affirmée devant :
La classification des épilepsies, définie par l’International League Against Epilepsy (ILAE) en 2017, permet une démarche synthétique réunissant le type d’épilepsie, le syndrome épileptique, l’étiologie ainsi que les comorbidités. Cette commission a également renommé certains syndromes épileptiques de l’enfant et précisé leurs critères diagnostiques principaux, en 2022.
Le diagnostic syndromique repose sur :
- la survenue de plusieurs crises épileptiques spontanées (au moins deux séparées de 24 heures) ;
- la survenue d’une crise spontanée dont le bilan étiologique permet de prédire que le risque de récidive est élevé ;
- la survenue de crise(s) d’épilepsie dont le bilan permet de poser le diagnostic d’un syndrome épileptique donné.
La classification des épilepsies, définie par l’International League Against Epilepsy (ILAE) en 2017, permet une démarche synthétique réunissant le type d’épilepsie, le syndrome épileptique, l’étiologie ainsi que les comorbidités. Cette commission a également renommé certains syndromes épileptiques de l’enfant et précisé leurs critères diagnostiques principaux, en 2022.
Le diagnostic syndromique repose sur :
- le type de crises prédominant, le contexte clinique, l’âge de début, l’examen clinique, les antécédents neurologiques personnels et familiaux, le développement ;
- les résultats de l’EEG, qui doit être réalisé le plus précocement possible après une première crise. Si l’EEG standard est normal, un EEG de longue durée ou un EEG de sommeil de sieste (jeune enfant) ou de nuit peuvent être réalisés afin de rechercher des anomalies intercritiques, ou enregistrer des crises. Un diagnostic d’épilepsie peut être affirmé avec un EEG normal dans certains cas ; un EEG anormal n’est pas forcément pathognomonique d’une épilepsie.
- l'imagerie par résonance magnétique (IRM) cérébrale, examen de référence, dont la lecture doit être orientée. La tomodensitométrie (TDM) cérébrale, moins précise, peut être utile lors d’une première crise pour rechercher une lésion nécessitant une prise en charge urgente ;
- des analyses génétiques qui sont proposées en cas de troubles du neurodéveloppement associés, sans anomalie structurelle identifiée. En complément des analyses chromosomiques, les nouvelles techniques de séquençage de haut débit permettent d’identifier des variations dans des gènes du développement cérébral ou de la neurotransmission, jusque dans 60 % des cas d’encéphalopathie épileptique et développementale à début précoce. Plus rarement, certaines épilepsies focales et généralisées familiales ont une origine monogénique ;
- des analyses immunologiques ou inflammatoires, en cas de début explosif de l’épilepsie ou en cas d’association à des troubles psychiatriques récents ;
- la recherche d’une cause métabolique ou une carence vitaminique, dans certains cas.
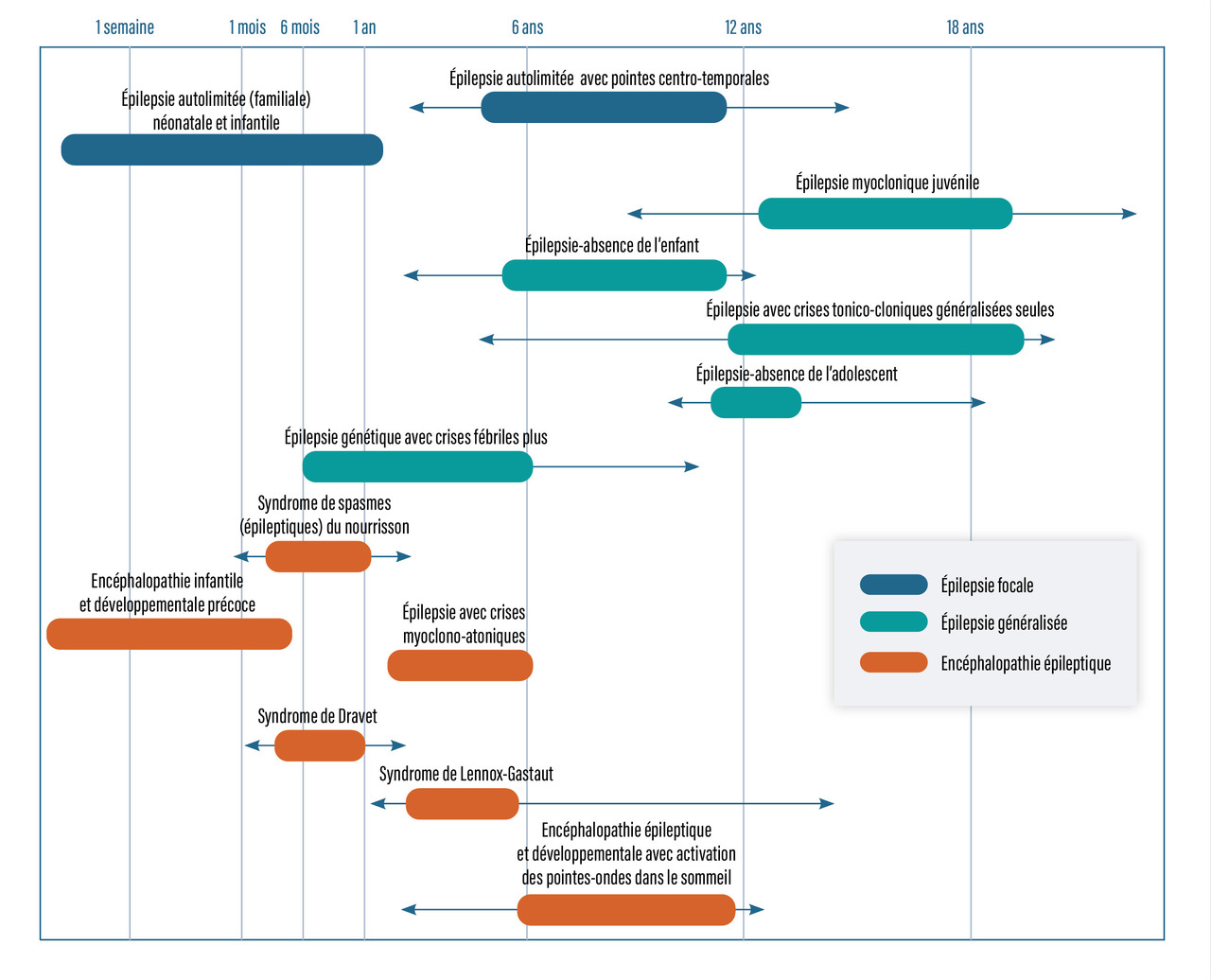
Principaux syndromes épileptiques de l’enfant
La figure 2 recense les différents syndromes épileptiques en fonction de l’âge de l’enfant.
Nourrisson
La survenue de crises provoquées chez le nourrisson est fréquente, mais le diagnostic d’épilepsie doit être évoqué devant la répétition de crises sans facteur déclenchant.
L’examen clinique peut être normal ou révéler des anomalies en lien avec la cause.
L’EEG peut montrer des éléments de focalisation (pointes ou ondes lentes) et l’EEG-vidéo prolongé peut parfois enregistrer une crise.
Les examens étiologiques recherchent une anomalie structurelle ou, plus rarement, une maladie génétique.
Un traitement médicamenteux est nécessaire, adapté à l’âge du nourrisson, pour plusieurs années.
Une forme particulière d’épilepsie du nourrisson avec crises focales, l’épilepsie infantile auto-limitée, est caractérisée par la survenue de crises focales en salve sur plusieurs jours, sans facteur déclenchant, parfois en contexte familial, chez un nourrisson sans antécédent personnel, au bon développement. Une cause génétique est fréquemment retrouvée. L’évolution est marquée par la guérison en quelques semaines. Ce diagnostic ne peut être affirmé qu’après avoir exclu les causes de crises provoquées ou une cause structurelle.
Les crises comportent des contractions des muscles axiaux de moins de trois secondes, en flexion ou en extension, répétées en série pendant plusieurs minutes, le plus souvent au réveil. Elles sont régulièrement confondues avec des manifestations digestives.
L’examen clinique peut être normal ou révéler des anomalies en lien avec la cause. Une régression psychomotrice, avec perte du contact social, est souvent observée.
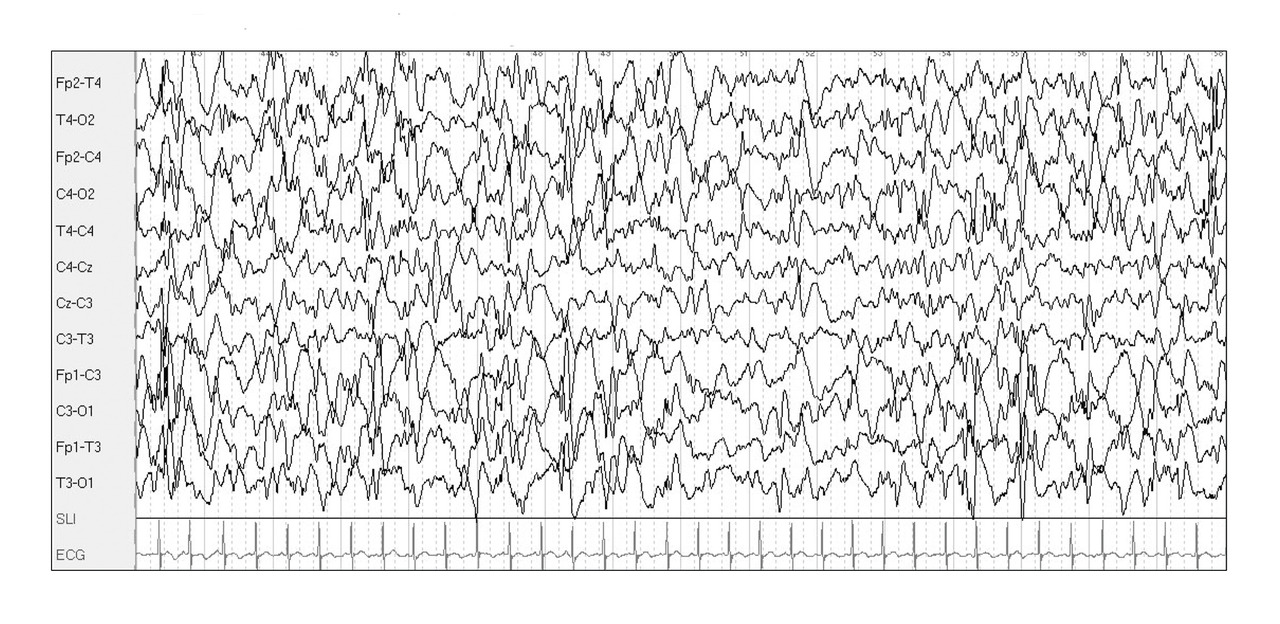
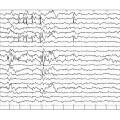
L’EEG montre une hypsarythmie (fig. 3 ), caractérisée par une disparition du rythme de fond et une désorganisation complète de l’activité électrique cérébrale, avec des ondes lentes et pointes multifocales diffuses et asynchrones de grande amplitude.
La recherche étiologique est importante : lésion cérébrale acquise précoce, malformation cérébrale, anomalie chromosomique, monogénique, sclérose tubéreuse de Bourneville, maladie métabolique…
Le traitement fait appel au vigabatrin, puis aux corticoïdes à forte dose en cas d’échec. Cette épilepsie est résistante aux divers traitements dans plus d’un tiers des cas.
Le pronostic développemental est conditionné par la durée d’évolution des spasmes, la réponse au traitement et la cause. Cette épilepsie peut guérir, ou évoluer vers une épilepsie focale, ou encore vers un syndrome de Lennox-Gastaut, avec déficit intellectuel et troubles autistiques.
Elles peuvent également entrer dans le cadre d’une épilepsie génétique avec crises fébriles plus (syndrome GEFS+ [generalized epilepsy with febrile seizures plus]).
Les crises fébriles peuvent débuter avant l’âge de 1 an et persister après 6 ans, et être associées à des crises non fébriles tonico-cloniques, jusqu’à l’âge adulte.
L’examen neurologique et le développement sont normaux, avec des antécédents familiaux fréquents.
L’EEG retrouve un rythme de fond normal avec parfois des pointes-ondes focales ou généralisées intercritiques. Près de 10 % des familles atteintes ont des mutations dans le gène SCN1A.
Le traitement médicamenteux est nécessaire, en évitant les molécules aggravantes, accompagné d’une éducation de la famille face aux crises.
Une forme rare et sévère, le syndrome de Dravet, est caractérisée par la survenue d’états de mal fébriles hémicorporels avant l’âge de 1 an, puis de crises non fébriles, évoluant vers une pharmacorésistance et un déficit intellectuel progressif.
Épilepsie avec crises focales
Les crises épileptiques focales du nourrisson sont difficiles à diagnostiquer, et souvent qualifiées de « malaises ».L’examen clinique peut être normal ou révéler des anomalies en lien avec la cause.
L’EEG peut montrer des éléments de focalisation (pointes ou ondes lentes) et l’EEG-vidéo prolongé peut parfois enregistrer une crise.
Les examens étiologiques recherchent une anomalie structurelle ou, plus rarement, une maladie génétique.
Un traitement médicamenteux est nécessaire, adapté à l’âge du nourrisson, pour plusieurs années.
Une forme particulière d’épilepsie du nourrisson avec crises focales, l’épilepsie infantile auto-limitée, est caractérisée par la survenue de crises focales en salve sur plusieurs jours, sans facteur déclenchant, parfois en contexte familial, chez un nourrisson sans antécédent personnel, au bon développement. Une cause génétique est fréquemment retrouvée. L’évolution est marquée par la guérison en quelques semaines. Ce diagnostic ne peut être affirmé qu’après avoir exclu les causes de crises provoquées ou une cause structurelle.
Syndrome des spasmes infantiles (anciennement syndrome de West)
Ce syndrome épileptique apparaît entre 1 et 24 mois, et constitue une véritable « encéphalopathie épileptique », ce qui en fait une urgence thérapeutique.Les crises comportent des contractions des muscles axiaux de moins de trois secondes, en flexion ou en extension, répétées en série pendant plusieurs minutes, le plus souvent au réveil. Elles sont régulièrement confondues avec des manifestations digestives.
L’examen clinique peut être normal ou révéler des anomalies en lien avec la cause. Une régression psychomotrice, avec perte du contact social, est souvent observée.
L’EEG montre une hypsarythmie (
La recherche étiologique est importante : lésion cérébrale acquise précoce, malformation cérébrale, anomalie chromosomique, monogénique, sclérose tubéreuse de Bourneville, maladie métabolique…
Le traitement fait appel au vigabatrin, puis aux corticoïdes à forte dose en cas d’échec. Cette épilepsie est résistante aux divers traitements dans plus d’un tiers des cas.
Le pronostic développemental est conditionné par la durée d’évolution des spasmes, la réponse au traitement et la cause. Cette épilepsie peut guérir, ou évoluer vers une épilepsie focale, ou encore vers un syndrome de Lennox-Gastaut, avec déficit intellectuel et troubles autistiques.
Épilepsies avec crises fébriles
Les crises fébriles concernent 2 à 5 % des enfants de moins de 5 ans et sont le plus souvent provoquées. Elles doivent faire immédiatement penser aux causes symptomatiques (infection du système nerveux, syndrome hémolytique et urémique…).Elles peuvent également entrer dans le cadre d’une épilepsie génétique avec crises fébriles plus (syndrome GEFS+ [generalized epilepsy with febrile seizures plus]).
Les crises fébriles peuvent débuter avant l’âge de 1 an et persister après 6 ans, et être associées à des crises non fébriles tonico-cloniques, jusqu’à l’âge adulte.
L’examen neurologique et le développement sont normaux, avec des antécédents familiaux fréquents.
L’EEG retrouve un rythme de fond normal avec parfois des pointes-ondes focales ou généralisées intercritiques. Près de 10 % des familles atteintes ont des mutations dans le gène SCN1A.
Le traitement médicamenteux est nécessaire, en évitant les molécules aggravantes, accompagné d’une éducation de la famille face aux crises.
Une forme rare et sévère, le syndrome de Dravet, est caractérisée par la survenue d’états de mal fébriles hémicorporels avant l’âge de 1 an, puis de crises non fébriles, évoluant vers une pharmacorésistance et un déficit intellectuel progressif.
Enfant
Épilepsie-absence de l’enfant
Cette épilepsie se manifeste entre 4 et 10 ans chez un enfant sans antécédent particulier, au bon développement initial. Ce syndrome, de cause inconnue, ou idiopathique, correspond à 10 % des épilepsies de l’enfant.Les absences épileptiques sont caractérisées par des ruptures de contact pluriquotidiennes durant de 3 à 20 secondes avec regard fixe, arrêt des activités, accompagnées parfois de clonies palpébrales, d’automatismes oraux ou manuels.
L’examen clinique est normal. Des difficultés d’apprentissage et/ou un trouble déficitaire de l’attention avec hyperactivité (TDAH) peuvent être retrouvés.
L’EEG-vidéo permet d’enregistrer des absences concomitantes de pointes-ondes généralisées rythmiques à 3 cycles par seconde caractéristiques, déclenchées par l’hyperpnée, parfois par la stimulation lumineuse intermittente, sur un rythme de fond normal.
Le pronostic est variable : pharmacosensibilité et guérison avant 10 ans dans deux tiers des cas, pharmacorésistance et évolution vers une épilepsie-absence de l’adolescent avec crises tonico-cloniques généralisées dans un tiers des cas.
Épilepsie (autolimitée) avec pointes centro-temporales
Ce syndrome épileptique fréquent (15 à 25 % des épilepsies de l’enfant) survient chez l’enfant âgé de 3 à 12 ans, sans antécédent neurologique, le plus souvent (rares antécédents de crises fébriles).Les crises focales sensitivo-motrices impliquent une hémiface ou un hémicorps, sont parfois secondairement généralisées, et de durée brève (moins de 2 à 3 minutes). Une hypersialorrhée puis une dysarthrie, ou un déficit moteur unilatéral transitoire (paralysie de Todd) peuvent être observés. Les crises sont favorisées par le sommeil, une heure après l’endormissement ou une à deux heures avant ou après le réveil. L’évolution vers un état de mal est exceptionnelle.
L’examen clinique est normal. Des difficultés d’apprentissages et/ou un trouble déficitaire de l’attention avec hyperactivité (TDAH) peuvent être retrouvés.
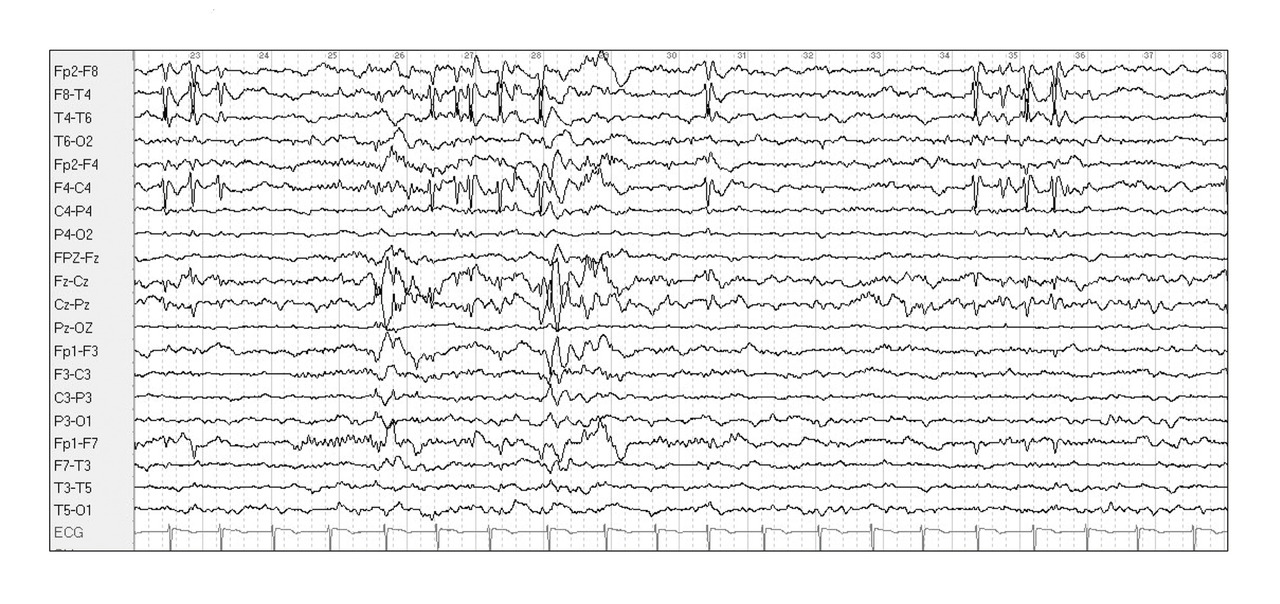
L’EEG intercritique de veille et de sommeil retrouve la présence de pointes diphasiques et parfois d’ondes lentes dans les régions centro-temporales, parfois bilatérales asynchrones, activées au sommeil. Ces anomalies caractéristiques permettent un diagnostic syndromique dès la première crise (
En cas de diagnostic électro-clinique certain, il n’y a pas d’indication à réaliser une IRM cérébrale si l’examen est normal.
Au vu du faible risque de récurrence de crises et de l’évolution spontanément favorable, avec guérison avant l’adolescence, l’abstention thérapeutique est proposée.
Dans environ 20 % des cas, l’épilepsie est plus active, avec crises fréquentes, parfois diurnes, avec absences et chutes atoniques, forte activation et diffusion des pointes sur l’EEG de sommeil, et retentissement cognitif et comportemental. Un traitement et un suivi rapproché doivent alors être proposés en évitant les médicaments aggravants.
Épilepsie symptomatique avec crises focales
La sémiologie des crises épileptiques focales varie en fonction de la topographie de la zone épileptogène. Les symptômes subjectifs sont difficiles à retrouver chez un enfant sans langage.L’examen clinique peut être normal ou révéler des anomalies en lien avec la cause.
L’EEG-vidéo permet de retrouver parfois un foyer d’ondes lentes ou de pointes en période postcritique ou intercritique.
La recherche étiologique est nécessaire avec, en premier lieu, la réalisation d’une IRM cérébrale, orientée par les informations cliniques. Elle est complétée en fonction des résultats et des éléments cliniques.
Un traitement est nécessaire, avec un contrôle possible des crises par une monothérapie, parfois une bithérapie, poursuivie en général jusqu’à l’âge adulte. Certaines causes structurelles malformatives ou post-infectieuses sont souvent associées à une pharmacorésistance (dysplasie corticale focale, tumeurs gliales de bas grade, lésions post-infectieuses...) et peuvent bénéficier d’un traitement chirurgical.
Adolescent
Épilepsies généralisées idiopathiques
Ces épilepsies débutent chez l’adolescent, sans antécédent personnel notable, entre 12 et 17 ans, et se poursuivent jusqu’à l’âge adulte. Elles sont de cause inconnue, dites « idiopathiques ».Certains aspects électro-cliniques sont communs :
- les premières crises sont généralisées tonico-cloniques, parfois favorisées par une dette de sommeil ;
- l’examen est normal. Il existe fréquemment des antécédents familiaux d’épilepsie ou de crises fébriles ;
- l’EEG intercritique retrouve un rythme de fond normal, des décharges de pointes-ondes ou polypointes-ondes généralisées entre 2,5 et 6 Hz, parfois déclenchées par l’hyperventilation ou la stimulation lumineuse intermittente ;
- un traitement est nécessaire afin d’éviter le risque d’accident, avec un bon contrôle des crises, dans la plupart des cas. Néanmoins, les restrictions de prescription du valproate de sodium peuvent rendre plus difficile le contrôle de cette épilepsie chez l’adolescente.
L’épilepsie myoclonique juvénile (EMJ) comporte également des myoclonies : secousses musculaires brèves en salve, asynchrones, essentiellement des membres supérieurs, survenant surtout le matin. Certains patients peuvent aussi présenter des absences (30 %). L’EEG retrouve des pointes-ondes généralisées, et parfois une photosensibilité.
Épilepsies focales
Les symptômes critiques et la démarche clinique, étiologique et thérapeutique chez l’adolescent sont superposables à ceux de l’enfant ou l’adulte.Toutefois, le diagnostic différentiel de crise psychogène non épileptique peut être difficile à cet âge, et l’EEG-vidéo de longue durée est parfois nécessaire.
Prise en charge thérapeutique
La prise en charge thérapeutique comporte plusieurs volets : le traitement d’urgence, le traitement au long cours, lorsqu’il est nécessaire, la prise en charge des comorbidités et l’éducation thérapeutique de l’enfant et de son entourage.
Traitement d’urgence
Situations d’urgence
Certaines crises épileptiques peuvent entraîner des conséquences vitales, en raison de leur survenue brutale, des symptômes dysautonomiques, associés à des troubles de la conscience :- les crises tonico-cloniques généralisées ;
- les crises prolongées avec vomissements, manifestations dysautonomiques, cyanose, apnées, bradycardie, asystolie ;
- l’état de mal épileptique, défini par la survenue d’une crise prolongée de plus de cinq minutes, ou de crises discontinues de plus de trente minutes. Il peut entraîner des conséquences hémodynamiques, et des modifications cérébrales vasculaires ou inflammatoires.
Les crises épileptiques avec chute ou perte de connaissance brutale, survenant sans prodromes, peuvent engendrer des accidents traumatiques, noyades, brûlures, accidents de la voie publique, en fonction du lieu et des circonstances de survenue. Il existe un risque de mort subite de 0,1 %, chez l’enfant ou l’adolescent avec crises généralisées nocturnes répétées, mal contrôlées par les traitements. Une information est nécessaire pour prévenir ces accidents.
Certains facteurs peuvent déstabiliser une épilepsie chez l’enfant, tels qu’une infection ORL ou digestive, un sevrage médicamenteux, un manque de sommeil ou de la fièvre. Un contact avec le neuropédiatre ou le neurologue doit alors être pris rapidement. L’administration ponctuelle d’une benzodiazépine orale peut être utile.
Prise en charge d’une crise d’épilepsie
Les crises brèves focales ou sans perte de connaissance avec retour rapide aux activités antérieures ne nécessitent pas de traitement d’urgence.En cas de crise généralisée tonico-clonique, une prise en charge urgente est indispensable pour sécuriser l’enfant :
- noter l’heure du début de la crise ;
- protéger l’enfant et l’allonger en position latérale de sécurité ;
- dégager les voies aériennes supérieures sans canule de Guedel ;
- si la crise dure plus de cinq minutes, administrer une benzodiazépine d’action rapide : diazépam administré par voie rectale (0,5 mg/kg, dose maximale 10 mg) ou midazolam gel buccal (0,3 mg/kg, dose maximale 10 mg) ;
- en cas de crise persistante au terme de dix minutes, une seconde dose de benzodiazépine doit être administrée, de préférence par voie intraveineuse en milieu hospitalier (urgences pédiatriques) ;
- un état de mal épileptique nécessite une prise en charge réanimatoire (Samu puis réanimation pédiatrique) avec traitement intraveineux (phénytoïne, lévétiracétam, phénobarbital).
Argumenter l’attitude thérapeutique et planifier le suivi du patient
L’introduction d’un traitement dépend du diagnostic syndromique et étiologique de l’épilepsie mais aussi du profil du patient. Certaines épilepsies liées à l’âge ne justifient pas de traitement au long cours, d’autres au contraire nécessitent un traitement rapidement efficace, en raison de la fréquence des crises et de la sévérité de leur retentissement développemental. Si la plupart des épilepsies de l’enfant sont pharmacosensibles, 25 % d’entre elles restent pharmacorésistantes dont un petit nombre seulement peuvent être guéries chirurgicalement.
Les traitements les plus utilisés chez l’enfant sont le valproate de sodium et la carbamazépine, le lévétiracétam et la lamotrigine. Leur introduction se fait en général progressivement, par paliers, pour améliorer la tolérance et éviter les réactions allergiques.
Un suivi clinique périodique spécialisé par un neuropédiatre ou un neurologue est nécessaire, afin d’évaluer l’observance, l’efficacité, la tolérance du traitement. Des effets indésirables somatiques (modification du comportement alimentaire, allergies), comportementaux, peuvent être observés et doivent être bien connus. Un suivi biologique annuel est nécessaire pour certains médicaments tels que le valproate de sodium, la carbamazépine (hémogramme, bilan hépatique).
En cas de pharmacorésistance, définie par la persistance de crises malgré deux monothérapies et une bithérapie bien conduites, une réévaluation complète de l’épilepsie en centre spécialisé est nécessaire. Des alternatives au traitement médicamenteux peuvent être proposées dans certaines situations telles que le traitement chirurgical curatif ou palliatif comme le stimulateur du nerf vague, ou le régime cétogène.
Les traitements les plus utilisés chez l’enfant sont le valproate de sodium et la carbamazépine, le lévétiracétam et la lamotrigine. Leur introduction se fait en général progressivement, par paliers, pour améliorer la tolérance et éviter les réactions allergiques.
Un suivi clinique périodique spécialisé par un neuropédiatre ou un neurologue est nécessaire, afin d’évaluer l’observance, l’efficacité, la tolérance du traitement. Des effets indésirables somatiques (modification du comportement alimentaire, allergies), comportementaux, peuvent être observés et doivent être bien connus. Un suivi biologique annuel est nécessaire pour certains médicaments tels que le valproate de sodium, la carbamazépine (hémogramme, bilan hépatique).
En cas de pharmacorésistance, définie par la persistance de crises malgré deux monothérapies et une bithérapie bien conduites, une réévaluation complète de l’épilepsie en centre spécialisé est nécessaire. Des alternatives au traitement médicamenteux peuvent être proposées dans certaines situations telles que le traitement chirurgical curatif ou palliatif comme le stimulateur du nerf vague, ou le régime cétogène.
Prise en charge des comorbidités
Les épilepsies du nourrisson et de l’enfant peuvent interférer avec le développement psychomoteur, puis cognitif et comportemental, et parfois survenir sur un terrain neurologique ou de troubles neurodéveloppementaux préexistants.
Le suivi d’une épilepsie nécessite donc une évaluation régulière du développement et du comportement de l’enfant, et la mise en œuvre de guidance et remédiations éventuelles (orthophonie, psychomotricité), voire d’un suivi psychothérapique. Outre le suivi clinique et de l’EEG, une évaluation neuropsychologique est parfois nécessaire pour orienter la prise en charge rééducative.
Ce suivi doit permettre de favoriser l’insertion scolaire et sociale de l’enfant. Une scolarité en structure spécialisée est parfois nécessaire. L’orientation professionnelle de l’adolescent doit également être envisagée suffisamment tôt.
Le suivi d’une épilepsie nécessite donc une évaluation régulière du développement et du comportement de l’enfant, et la mise en œuvre de guidance et remédiations éventuelles (orthophonie, psychomotricité), voire d’un suivi psychothérapique. Outre le suivi clinique et de l’EEG, une évaluation neuropsychologique est parfois nécessaire pour orienter la prise en charge rééducative.
Ce suivi doit permettre de favoriser l’insertion scolaire et sociale de l’enfant. Une scolarité en structure spécialisée est parfois nécessaire. L’orientation professionnelle de l’adolescent doit également être envisagée suffisamment tôt.
Éducation thérapeutique
Comme pour toute maladie chronique, une éducation thérapeutique de l’enfant et des parents est nécessaire, abordant les connaissances sur la maladie, les traitements, la conduite à tenir en cas d’urgence, l’hygiène de vie, les facteurs de risque et les mesures de protection. Cette éducation nécessite des outils adaptés aux différentes tranches d’âge et aux capacités de compréhension de l’enfant. Elle est particulièrement importante à l’adolescence, et doit encore être développée dans le domaine de l’épilepsie.
Points forts
Épilepsie de l’enfant et de l’adulte
POINTS FORTS À RETENIR
Les épilepsies de l’enfant diffèrent par leur âge de survenue, pronostic et étiologie.
Les épilepsies dites « autolimitées » guérissent à l’adolescence.
La conduite à tenir devant une crise d’épilepsie repose sur les mesures de protection immédiate, la mesure de la durée de la crise et l’introduction d’un traitement d’urgence par benzodiazépines en cas de crise de durée supérieure à cinq minutes.
Le suivi développemental et des apprentissages de l’enfant avec épilepsie est essentiel.
En cas de résistance au traitement antiépileptique, une réévaluation de l’épilepsie doit être réalisée en centre spécialisé.
Pour en savoir plus
Haute Autorité de santé. Guide du parcours de santé de l’enfant avec épilepsie. Mis en ligne le 14 juin 2023.
Haute Autorité de santé. Épilepsies. Prise en charge des enfants et des adultes. Recommandation de bonne pratique. Mis en ligne le 26 novembre 2020.
https://www.epilepsydiagnosis.org/
https://www.ilae.org/
https://www.epilepsie-info.fr/
Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1398-442.
Scheffer IE, Berkovic S, Capovilla G, Connoly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512-21.
Haute Autorité de santé. Épilepsies. Prise en charge des enfants et des adultes. Recommandation de bonne pratique. Mis en ligne le 26 novembre 2020.
https://www.epilepsydiagnosis.org/
https://www.ilae.org/
https://www.epilepsie-info.fr/
Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1398-442.
Scheffer IE, Berkovic S, Capovilla G, Connoly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512-21.
 Encadrés
Encadrés