Une crise d’épilepsie chez un enfant est un événement traumatisant pour les parents sur le plan émotionnel qui engendre une anxiété face à la survenue d’une récurrence. Il faut leur expliquer les risques réels, s’accorder sur la conduite à tenir en cas de récidive, et limiter les modifications éducatives inappropriées.
L’épilepsie est un groupe de maladies fréquentes et touche 1 personne sur 26 tout au long de la vie. La prévalence en France est de 1/1 000 et touche 200 000 à 300 000 enfants en France. On doit plutôt parler des épilepsies que de l’épilepsie, car il existe un grand nombre d’épilepsies différentes. Un diagnostic précis permet d’expliquer la maladie, de choisir un traitement adapté et d’évoquer le pronostic. La Ligue internationale contre l’épilepsie (International League Against Epilepsy [ILAE]) définit la crise épileptique comme une manifestation transitoire de signes et/ou de symptômes due à une activité neuronale cérébrale anormalement excessive ou synchrone.1 La crise épileptique est un symptôme, tandis que les épilepsies sont des maladies du cerveau caractérisées par une prédisposition durable à générer des crises d’épilepsie, avec ses conséquences neurobiologiques, cognitives, psychologiques et sociales.1 L’épilepsie est une maladie du cerveau définie par l’une des trois conditions suivantes : au moins deux crises non provoquées (ou réflexes) survenant à plus de 24 heures d’intervalle ; une crise non provoquée (ou réflexe) et une probabilité d’autres crises semblables avec un risque général de récurrence d’au moins 60 % survenant au cours des 10 prochaines années ; le diagnostic d’un syndrome épileptique.1
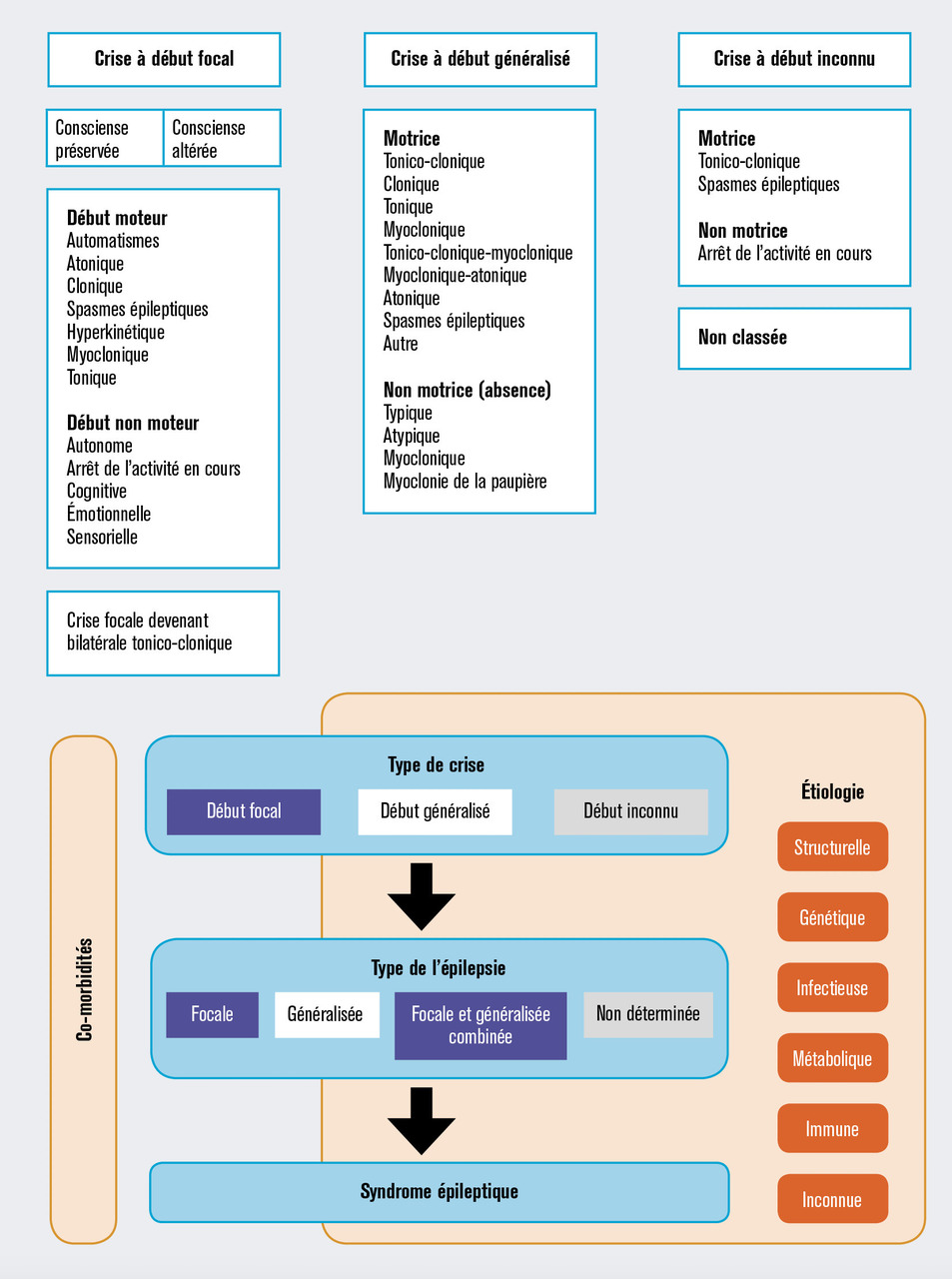
Classification des crises et syndromes épileptiques
Les différents types de crises ainsi que les différentes formes d’épilepsies sont décrits par des classifications de l’ILAE ; les classifications des types de crises et de l’organisation des épilepsies ont été révisées en 2017.2, 3
Les crises d’épilepsie se divisent en crises à début focal, à début généralisé ou à début inconnu, avec des sous-catégories de crises motrices ou non motrices et avec ou sans modification de la conscience pour les crises focales.3 La classification des crises d’épilepsie commence par déterminer si les manifestations initiales des crises sont focales ou généralisées. Le début de la crise peut avoir été manqué ou ne pas être clair, auquel cas la crise est considérée à début inconnu (fig. 1 ).3
Les crises à début focal peuvent évoluer avec une implication de réseaux cérébraux bilatéraux, et on parle alors de crise d’épilepsie « focale devenant bilatérale tonico-clonique ». Ces crises focales devenant bilatérales tonico-cloniques représentent un modèle de propagation d’une crise d’épilepsie plutôt qu’un type de crise unitaire. Par le passé, on parlait de « crise à début partiel avec crise secondairement généralisée ». Les termes « devenant bilatérale » remplaçant les termes « secondairement généralisée ». Le terme « bilatéral » désigne le mode de propagation alors que le terme « généralisée » est utilisé pour les crises d’épilepsie qui impliquent des réseaux bilatéraux dès le début. Les crises à début focal peuvent ne pas évoluer vers une bilatéralisation en impliquant seulement un réseau de neurones unilatéral.
Les épilepsies quant à elles s’organisent en trois niveaux (fig. 1 ) :2
– le premier niveau est la caractérisation du type de crise, fondé sur la classification des crises de 2017 décrite brièvement ci-dessus ;
– le deuxième niveau est la caractérisation du type d’épilepsie parmi les quatre types suivants : épilepsie focale, épilepsie généralisée, épilepsie généralisée et focale combinée, et épilepsie de type inconnu ;
– le troisième niveau est la détermination du syndrome épileptique spécifique quand cela est possible.
La classification des épilepsies intègre la cause à chaque niveau pour souligner la nécessité de la considérer à chaque étape du diagnostic, car elle comporte souvent des implications thérapeutiques importantes. L’étiologie est divisée en six sous-groupes : génétique, structurelle, métabolique, immune, infectieuse et inconnue. De plus, les comorbidités du fait de leurs fréquences sont également à considérer à chaque niveau diagnostique. C’est pour cette raison qu’elles apparaissent dans l’organisation des épilepsies.2
Les crises d’épilepsie se divisent en crises à début focal, à début généralisé ou à début inconnu, avec des sous-catégories de crises motrices ou non motrices et avec ou sans modification de la conscience pour les crises focales.3 La classification des crises d’épilepsie commence par déterminer si les manifestations initiales des crises sont focales ou généralisées. Le début de la crise peut avoir été manqué ou ne pas être clair, auquel cas la crise est considérée à début inconnu (
Les crises à début focal peuvent évoluer avec une implication de réseaux cérébraux bilatéraux, et on parle alors de crise d’épilepsie « focale devenant bilatérale tonico-clonique ». Ces crises focales devenant bilatérales tonico-cloniques représentent un modèle de propagation d’une crise d’épilepsie plutôt qu’un type de crise unitaire. Par le passé, on parlait de « crise à début partiel avec crise secondairement généralisée ». Les termes « devenant bilatérale » remplaçant les termes « secondairement généralisée ». Le terme « bilatéral » désigne le mode de propagation alors que le terme « généralisée » est utilisé pour les crises d’épilepsie qui impliquent des réseaux bilatéraux dès le début. Les crises à début focal peuvent ne pas évoluer vers une bilatéralisation en impliquant seulement un réseau de neurones unilatéral.
Les épilepsies quant à elles s’organisent en trois niveaux (
– le premier niveau est la caractérisation du type de crise, fondé sur la classification des crises de 2017 décrite brièvement ci-dessus ;
– le deuxième niveau est la caractérisation du type d’épilepsie parmi les quatre types suivants : épilepsie focale, épilepsie généralisée, épilepsie généralisée et focale combinée, et épilepsie de type inconnu ;
– le troisième niveau est la détermination du syndrome épileptique spécifique quand cela est possible.
La classification des épilepsies intègre la cause à chaque niveau pour souligner la nécessité de la considérer à chaque étape du diagnostic, car elle comporte souvent des implications thérapeutiques importantes. L’étiologie est divisée en six sous-groupes : génétique, structurelle, métabolique, immune, infectieuse et inconnue. De plus, les comorbidités du fait de leurs fréquences sont également à considérer à chaque niveau diagnostique. C’est pour cette raison qu’elles apparaissent dans l’organisation des épilepsies.2
Généralités sur les épilepsies de l’enfant
La survenue d’une crise épileptique est souvent un événement traumatisant pour le patient et sa famille. Si les parents ne l’expriment pas spontanément, ils ont le plus souvent vécu un épisode traumatisant sur le plan émotionnel, avec un sentiment intense de danger imminent ou de décès de leur enfant. Cela engendre une anxiété face à la survenue d’une récurrence. Il est donc fondamental que les soignants prennent le temps d’écouter et d’expliquer ce qui s’est passé et ce qui peut se passer. Il faut dire aux parents que leur ressenti est identique à celui de nombreux parents. Il faut, en revanche, expliquer les risques réels des crises épileptiques, s’accorder sur la conduite à tenir en cas de récidive et limiter les modifications éducatives inappropriées.4
Face à la peur de récurrence des crises, les parents et l’entourage ont tendance à modifier leurs attitudes éducatives et les habitudes de vie de leur enfant. Il n’est pas rare que cela résulte en un certain nombre d’interdictions et de restrictions dans l’autonomie. Par exemple, cela peut se manifester chez les enfants les plus jeunes par une surveillance accrue, ou par des modifications dans les habitudes du coucher ou la mise en place d’une surveillance pendant le sommeil. Chez les plus grands, l’utilisation des écrans (téléphone, tablette, ordinateur, télévision), en particulier pour les jeux vidéo, est très souvent limitée ou interdite par peur d’apparition d’une crise épileptique, même si, en pratique, les crises photo-induites sont très rares. Il n’y a donc pas lieu que les parents modifient leur choix éducatif concernant ces activités. Concernant la pratique sportive, il faut évaluer le maintien ou la restriction en fonction du type de sport, du type d’épilepsie, du traitement et de la réponse au traitement. Il n’y pas de contre-indication médicale systématique pour le grimper de corde et les activités en piscine même si ces activités cristallisent les craintes de l’entourage. La baignade avec une surveillance adaptée est parfaitement possible chez une grande majorité des patients, en faisant preuve d’une prudence transitoire pour les patients en début de traitement ou avec une accélération de la fréquence des crises.
Face à la peur de récurrence des crises, les parents et l’entourage ont tendance à modifier leurs attitudes éducatives et les habitudes de vie de leur enfant. Il n’est pas rare que cela résulte en un certain nombre d’interdictions et de restrictions dans l’autonomie. Par exemple, cela peut se manifester chez les enfants les plus jeunes par une surveillance accrue, ou par des modifications dans les habitudes du coucher ou la mise en place d’une surveillance pendant le sommeil. Chez les plus grands, l’utilisation des écrans (téléphone, tablette, ordinateur, télévision), en particulier pour les jeux vidéo, est très souvent limitée ou interdite par peur d’apparition d’une crise épileptique, même si, en pratique, les crises photo-induites sont très rares. Il n’y a donc pas lieu que les parents modifient leur choix éducatif concernant ces activités. Concernant la pratique sportive, il faut évaluer le maintien ou la restriction en fonction du type de sport, du type d’épilepsie, du traitement et de la réponse au traitement. Il n’y pas de contre-indication médicale systématique pour le grimper de corde et les activités en piscine même si ces activités cristallisent les craintes de l’entourage. La baignade avec une surveillance adaptée est parfaitement possible chez une grande majorité des patients, en faisant preuve d’une prudence transitoire pour les patients en début de traitement ou avec une accélération de la fréquence des crises.
Épilepsies les plus fréquentes chez l’enfant
Épilepsie avec crises focales
Les épilepsies avec crises focales représentant 50 à 60 % des épilepsies de l’enfant. Il s’agit d’un groupe très hétérogène. On ne peut donc pas parler de syndrome. Ces épilepsies peuvent débuter à n’importe quel âge, avec une symptomatologie qui varie en fonction du point de départ des crises. Il peut y avoir différentes causes, et la réponse au traitement peut être très variable.
Le diagnostic est suspecté le plus souvent sur l’interrogatoire qui permet de décrire la sémiologie des crises, mais cela est parfois difficile. Les signes de focalisation peuvent être brefs ou discrets ou rapidement suivis d’une évolution tonico-clonique bilatérale qui peut à tort faire évoquer des crises à début généralisé. Dans les premiers signes d’une crise à début focal, on peut retrouver des sensations anormales (nausées, paresthésies...), une déviation des yeux et/ou de la tête d’un côté, des clonies d’un membre ou d’un hémicorps... L’électro-encéphalogramme (EEG) intercritique peut être normal ou mettre en évidence un foyer épileptique. L’enregistrement est le plus souvent réalisé entre les crises. Il est le plus souvent normal, ce qui n’élimine pas le diagnostic. L’imagerie cérébrale par résonance magnétique (IRM) est indispensable pour rechercher une cause telle qu’une anomalie de développement du cortex ou une cicatrice gliale. Les tumeurs sont très rares mais sont parfois retrouvées. La normalité de l’IRM est plutôt un facteur de bon pronostic du contrôle des crises épileptiques.
Dans les épilepsies avec crises focales, il peut y avoir des comorbidités. Les plus fréquentes sont le trouble déficit de l’attention avec ou sans hyperactivité (TDAH) et les difficultés cognitives. Le TDAH concerne un tiers des épilepsies débutantes. Le plus souvent, il s’agit d’une forme inattentive pure. Les difficultés cognitives peuvent concerner un domaine cognitif spécifique ou l’ensemble des domaines. Un âge précoce de début d’épilepsie, des signes neurologiques ou une imagerie anormale sont des facteurs de risque pour une atteinte cognitive.
Le traitement est choisi en fonction de l’efficacité de chaque molécule, de son profil d’effets indésirables et du profil individuel de chaque patient.5
Le diagnostic est suspecté le plus souvent sur l’interrogatoire qui permet de décrire la sémiologie des crises, mais cela est parfois difficile. Les signes de focalisation peuvent être brefs ou discrets ou rapidement suivis d’une évolution tonico-clonique bilatérale qui peut à tort faire évoquer des crises à début généralisé. Dans les premiers signes d’une crise à début focal, on peut retrouver des sensations anormales (nausées, paresthésies...), une déviation des yeux et/ou de la tête d’un côté, des clonies d’un membre ou d’un hémicorps... L’électro-encéphalogramme (EEG) intercritique peut être normal ou mettre en évidence un foyer épileptique. L’enregistrement est le plus souvent réalisé entre les crises. Il est le plus souvent normal, ce qui n’élimine pas le diagnostic. L’imagerie cérébrale par résonance magnétique (IRM) est indispensable pour rechercher une cause telle qu’une anomalie de développement du cortex ou une cicatrice gliale. Les tumeurs sont très rares mais sont parfois retrouvées. La normalité de l’IRM est plutôt un facteur de bon pronostic du contrôle des crises épileptiques.
Dans les épilepsies avec crises focales, il peut y avoir des comorbidités. Les plus fréquentes sont le trouble déficit de l’attention avec ou sans hyperactivité (TDAH) et les difficultés cognitives. Le TDAH concerne un tiers des épilepsies débutantes. Le plus souvent, il s’agit d’une forme inattentive pure. Les difficultés cognitives peuvent concerner un domaine cognitif spécifique ou l’ensemble des domaines. Un âge précoce de début d’épilepsie, des signes neurologiques ou une imagerie anormale sont des facteurs de risque pour une atteinte cognitive.
Le traitement est choisi en fonction de l’efficacité de chaque molécule, de son profil d’effets indésirables et du profil individuel de chaque patient.5
Épilepsie à pointes centro-temporales
L’épilepsie à pointes centro-temporales représente 15 % des épilepsies de l’enfant.6 Il s’agit d’une épilepsie autolimitée. Cela veut dire que l’évolution naturelle de ce syndrome se fait vers la disparition des crises. Elle débute en moyenne entre 5 et 10 ans (2-14 ans). Les crises sont généralement en rapport avec le sommeil (après l’endormissement ou après le réveil) mais peuvent parfois survenir dans la journée. Elles sont caractérisées par des signes sensivito-moteurs comme des paresthésies au niveau buccal et des clonies faciales qui peuvent se propager au niveau des membres supérieurs et inférieurs. Il n’est pas rare qu’il y ait une aphasie. Certaines crises sont sans rupture de conscience alors que pour d’autres l’évolution se fait vers une rupture de conscience, voire vers une implication tonico-clonique bilatérale. Les crises sont en général brèves (1-2 min) et peu fréquentes (1-4 par an).
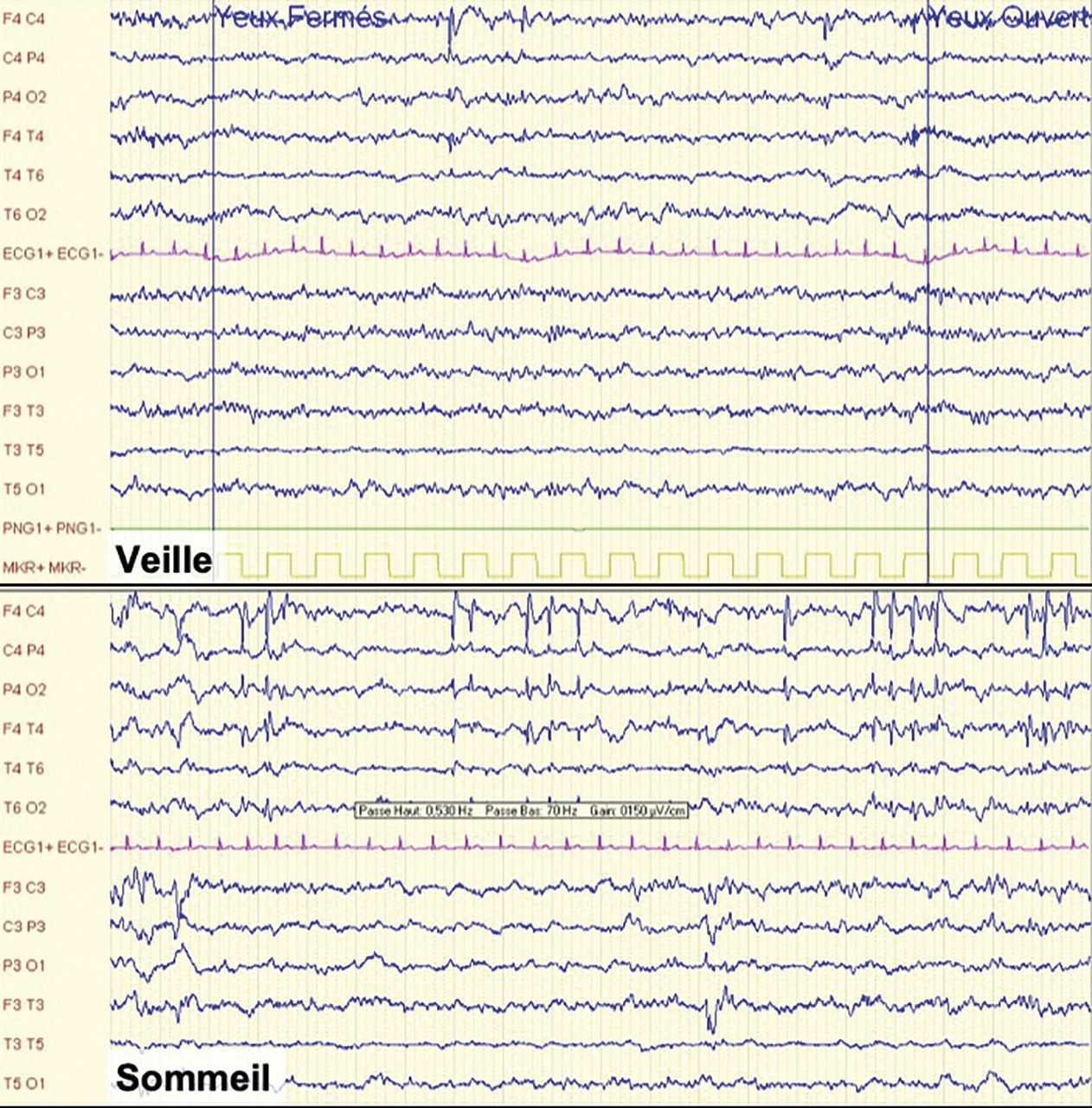
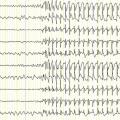
Il est rare d’enregistrer une crise, mais l’enregistrement de l’électroencéphalogramme (EEG) entre les crises (intercritique) associé à l’histoire clinique permet le diagnostic. Sur le tracé, on note des pointes biphasiques isolées ou groupées en bouffées dans les régions centro-temporales, d’un ou des deux côtés, accentuées par le sommeil (fig. 2 ). L’imagerie cérébrale n’est pas indiquée et est normale.
Les crises disparaissent avec ou sans traitement avant la fin de l’adolescence. Si les crises sont trop fréquentes, un traitement antiépileptique peut être instauré, en tenant compte des effets indésirables, en particulier sur le plan cognitif. L’évolution est favorable, mais il faut bien dépister les 10 à 30 % des patients ayant des comorbidités comme un TDAH, des difficultés de langage ou des difficultés praxiques. Il faut être particulièrement vigilant pour ceux cumulant plusieurs de ces comorbidités car le risque sur leur scolarité est le plus important. L’avis d’un spécialiste est souhaitable avant que les difficultés scolaires soient établies.
Il faut savoir remettre en cause le diagnostic d’épilepsie à pointes centro-temporales en cas de résistance aux traitements antiépileptiques, de récurrence trop fréquente des crises ou encore en cas de persistance de l’épilepsie à l’adolescence.
Une dégradation cognitive chez un enfant suivi pour une épilepsie à pointes centro-temporales doit conduire à revoir l’enfant et à réaliser un EEG en urgence afin de conduire rapidement au diagnostic et à la prise en charge d’une encéphalopathie épileptique avec pointes continues du sommeil.
Il est rare d’enregistrer une crise, mais l’enregistrement de l’électroencéphalogramme (EEG) entre les crises (intercritique) associé à l’histoire clinique permet le diagnostic. Sur le tracé, on note des pointes biphasiques isolées ou groupées en bouffées dans les régions centro-temporales, d’un ou des deux côtés, accentuées par le sommeil (
Les crises disparaissent avec ou sans traitement avant la fin de l’adolescence. Si les crises sont trop fréquentes, un traitement antiépileptique peut être instauré, en tenant compte des effets indésirables, en particulier sur le plan cognitif. L’évolution est favorable, mais il faut bien dépister les 10 à 30 % des patients ayant des comorbidités comme un TDAH, des difficultés de langage ou des difficultés praxiques. Il faut être particulièrement vigilant pour ceux cumulant plusieurs de ces comorbidités car le risque sur leur scolarité est le plus important. L’avis d’un spécialiste est souhaitable avant que les difficultés scolaires soient établies.
Il faut savoir remettre en cause le diagnostic d’épilepsie à pointes centro-temporales en cas de résistance aux traitements antiépileptiques, de récurrence trop fréquente des crises ou encore en cas de persistance de l’épilepsie à l’adolescence.
Une dégradation cognitive chez un enfant suivi pour une épilepsie à pointes centro-temporales doit conduire à revoir l’enfant et à réaliser un EEG en urgence afin de conduire rapidement au diagnostic et à la prise en charge d’une encéphalopathie épileptique avec pointes continues du sommeil.
Épilepsie-absence de l’enfant
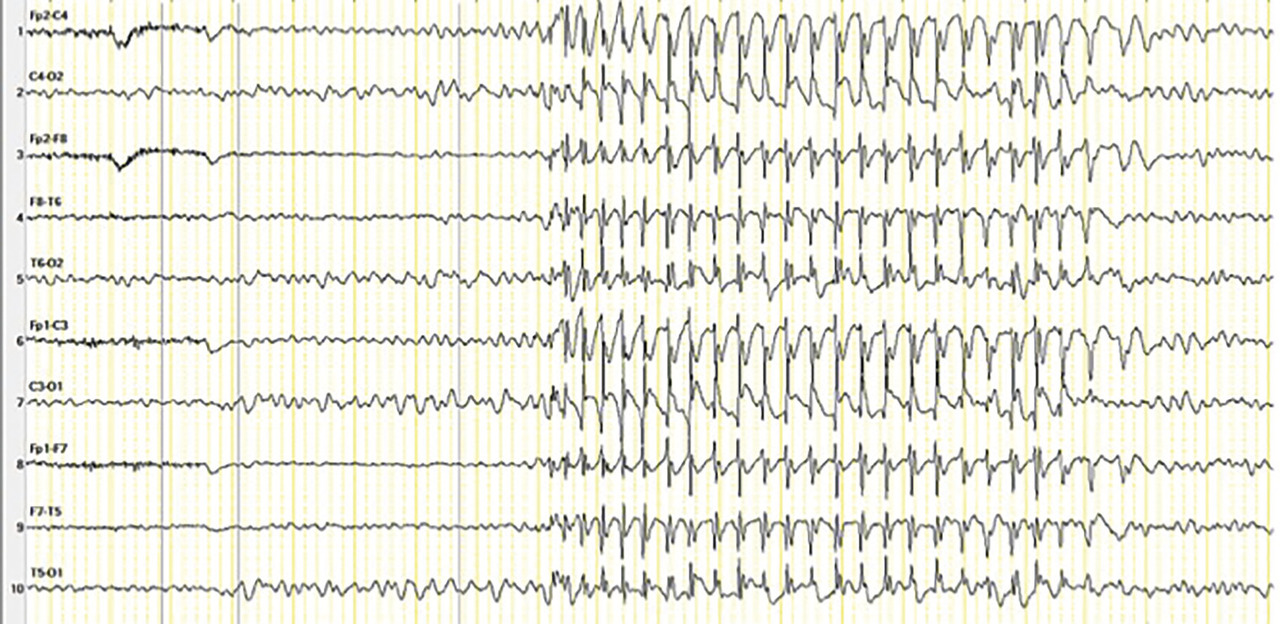
L’épilepsie-absence représente 10 % des épilepsies de l’enfant.7 Elle survient en moyenne entre 6 et 7 ans (4-10 ans). Les enfants ont un développement antérieur normal, avec parfois des troubles attentionnels préexistants. Dans ce syndrome, un seul type de crise est observé au moment du diagnostic : les crises d’absences typiques qui sont des ruptures de contact, de durée de 4 à 20 secondes (8 sec en moyenne), à début et fin brutal parfois accompagnées d’automatismes.7 Elles surviennent plusieurs dizaines de fois par jour. Leur enregistrement à l’EEG est indispensable, permettant d’identifier des pointes ondes généralisées à 3 Hz concomitantes à la rupture de contact (f ig. 3 ). Le diagnostic est fait en consultation (absences provoquées par l’hyperpnée) et confirmé par l’EEG. Aucun autre examen n’est nécessaire. Un avis spécialisé est nécessaire en cas de début précoce avant l’âge de 4 ans, en absence de réponse aux premières lignes de traitement ou en cas d’atteinte cognitive.7
Le pronostic de l’épilepsie-absence est le plus souvent favorable puisque dans 4 cas sur 5 le traitement va contrôler les absences, permettant un arrêt du traitement après 2 ans sans crise. Toutefois, chez ces patients contrôlés, entre 5 et 10 % auront une épilepsie ultérieure. Le traitement débute par une monothérapie par éthosuximide et en cas d’échec on utilise le valproate de sodium, puis la lamotrigine. L’objectif du traitement est qu’il n’y ait plus aucune absence et une bonne tolérance du médicament. Le dépistage d’un TDAH est systématique car ce trouble est observé dans sa forme inattentive chez 30 % des patients.7
Le pronostic de l’épilepsie-absence est le plus souvent favorable puisque dans 4 cas sur 5 le traitement va contrôler les absences, permettant un arrêt du traitement après 2 ans sans crise. Toutefois, chez ces patients contrôlés, entre 5 et 10 % auront une épilepsie ultérieure. Le traitement débute par une monothérapie par éthosuximide et en cas d’échec on utilise le valproate de sodium, puis la lamotrigine. L’objectif du traitement est qu’il n’y ait plus aucune absence et une bonne tolérance du médicament. Le dépistage d’un TDAH est systématique car ce trouble est observé dans sa forme inattentive chez 30 % des patients.7
Épilepsie myoclonique juvénile
L’épilepsie myoclonique juvénile débute aux alentours de 14 ans (8-26 ans). Elle représente entre 5 et 10 % de l’ensemble des épilepsies.8 Il existe différents types de crises dans cette épilepsie, avec des myoclonies épileptiques, des crises tonico-cloniques généralisées (peu fréquentes, survenant essentiellement le matin au réveil) et des crises d’absences typiques dans 30 % des cas.
Le diagnostic est le plus souvent porté à la suite de la première crise généralisée, avec la présence de myoclonies survenant essentiellement le matin au réveil évoluant souvent depuis 12 à 18 mois. L’EEG peut enregistrer des myoclonies, ou des anomalies généralisées sans manifestations cliniques à type de bouffées de pointes-ondes généralisées et peut retrouver une photosensibilité dans 30 % des cas.
Quand l’histoire clinique et l’EEG sont compatibles avec une épilepsie myoclonique juvénile, il n’y a pas lieu de réaliser d’autres examens complémentaires. En revanche, en cas d’anomalie de l’examen neurologique, de régression, de troubles du comportement et d’évolution défavorable de l’épilepsie, un avis spécialisé est nécessaire.
Le diagnostic est le plus souvent porté à la suite de la première crise généralisée, avec la présence de myoclonies survenant essentiellement le matin au réveil évoluant souvent depuis 12 à 18 mois. L’EEG peut enregistrer des myoclonies, ou des anomalies généralisées sans manifestations cliniques à type de bouffées de pointes-ondes généralisées et peut retrouver une photosensibilité dans 30 % des cas.
Quand l’histoire clinique et l’EEG sont compatibles avec une épilepsie myoclonique juvénile, il n’y a pas lieu de réaliser d’autres examens complémentaires. En revanche, en cas d’anomalie de l’examen neurologique, de régression, de troubles du comportement et d’évolution défavorable de l’épilepsie, un avis spécialisé est nécessaire.
Épilepsie-absence de l’adolescent
L’épilepsie-absence de l’adolescent débute aux alentours de 10-12 ans (7-17 ans). Elle représente 2 % de toutes les épilepsies, et est moins fréquente que l’épilepsie myoclonique juvénile.8
Ce syndrome est différent de l’épilepsie-absence de l’enfant, et cela a une implication importante sur le choix du traitement. Il existe deux types de crises, avec des crises-absences et des crises tonico-cloniques généralisées (peu fréquentes, survenant surtout le matin au réveil). En général, les absences sont moins nombreuses que dans l’épilepsie-absence de l’enfant et peuvent donc passées inaperçues. Le diagnostic est porté sur une analyse électro-clinique après la première crise généralisée ou devant la présence d’absences. L’EEG peut enregistrer des absences favorisées par une épreuve d’hyperpnée, ou des anomalies généralisées sans manifestations cliniques à type de bouffées de pointes-ondes généralisées. Quand l’histoire clinique et l’EEG sont compatibles avec une épilepsie-absence de l’ado-lescent, il n’y a pas lieu de réaliser d’autres examens complémentaires.
Ce syndrome est différent de l’épilepsie-absence de l’enfant, et cela a une implication importante sur le choix du traitement. Il existe deux types de crises, avec des crises-absences et des crises tonico-cloniques généralisées (peu fréquentes, survenant surtout le matin au réveil). En général, les absences sont moins nombreuses que dans l’épilepsie-absence de l’enfant et peuvent donc passées inaperçues. Le diagnostic est porté sur une analyse électro-clinique après la première crise généralisée ou devant la présence d’absences. L’EEG peut enregistrer des absences favorisées par une épreuve d’hyperpnée, ou des anomalies généralisées sans manifestations cliniques à type de bouffées de pointes-ondes généralisées. Quand l’histoire clinique et l’EEG sont compatibles avec une épilepsie-absence de l’ado-lescent, il n’y a pas lieu de réaliser d’autres examens complémentaires.
Prise en charge de l’épilepsie myoclonique juvénile et de l’épilepsie-absence de l’adolescent
L’objectif du traitement dans ces deux syndromes est que l’enfant ou l’adolescent soit libre de crise avec peu ou pas d’effets indésirables. Le traitement de fond est débuté dès le diagnostic et souvent après la première crise généralisée. Le choix du traitement se fait en fonction de la rapidité souhaitée pour la mise en place du traitement, du sexe et du profil du patient. Les principales molécules utilisées sont le lévétiracétam, la lamotrigine et le valproate de sodium. Ces molécules sont à la fois efficaces sur les crises tonico-cloniques généralisées mais aussi sur les myoclonies épileptiques et les crises-absences. Il faut savoir que le traitement sera probablement pris à vie du fait de la pharmacodépendance de ces deux syndromes. Le valproate est à éviter en première intention chez les jeunes femmes, mais il ne faut pas se priver de ce médicament, le plus efficace dans ces syndromes, en cas de non-réponse aux traitements de premières lignes. À noter que l’éthosuximide qui est le traitement de première intention de l’épilepsie-absence de l’enfant ne sera jamais prescrite en monothérapie dans l’épilepsie-absence de l’adolescent car elle n’est pas efficace sur les crises tonico-cloniques généralisées.
Dans ces syndromes, les crises sont favorisées par l’alcool, le manque de sommeil, la stimulation lumineuse… Une bonne hygiène de vie est aussi importante que l’observance thérapeutique.
Dans ces syndromes, les crises sont favorisées par l’alcool, le manque de sommeil, la stimulation lumineuse… Une bonne hygiène de vie est aussi importante que l’observance thérapeutique.
Une prise en charge personnalisée
En plus de la prise en charge médicamenteuse, il faut prendre en considération tout ce qui constitue la vie de l’enfant. C’est-à-dire sa vie au sein de sa famille, ses apprentissages et sa scolarité, et aussi ses activités extrascolaires. Il faut constamment dépister, prendre en charge et accompagner les enfants, les adolescents et leur famille dans l’évolution de leur maladie.
Le choix des antiépileptiques est fondé sur les données cliniques ayant démontré leur efficacité et sur la balance bénéfice-risque à l’échelle individuelle (v. tableau
L’objectif du traitement est différent d’un patient à un autre. Il dépend du syndrome et de l’évolution de la maladie. Chez les enfants ayant une épilepsie débutante, l’objectif thérapeutique est d’obtenir un contrôle complet des crises épileptiques sans effet indésirable afin de permettre la vie la plus normale possible à cet enfant. En revanche, chez les enfants avec une épilepsie résistante, l’objectif n’est plus nécessairement d’être libres de crises. Chez ces patients, on cherche plutôt à améliorer la qualité de vie en évitant la survenue des crises les plus gênantes (par exemple : disparition des crises focales avec conscience altérée ou avec évolution bilatérale tonico-clonique) avec un taux d’effets indésirables le plus léger possible.
La scolarité et les apprentissages occupent une grande part de la vie des enfants. Un peu plus de 50 % des enfants atteints d’épilepsie ont des troubles scolaires. Chaque consultation doit donc comprendre une partie de dépistage de ces difficultés. Il faut s’intéresser tant aux performances qu’aux moyens mis en place pour obtenir un tel résultat. En cas de difficultés scolaires, il faut informer les parents et le médecin scolaire. Une évaluation neuropsychologique peut permettre de mieux caractériser les difficultés pour mettre en place une stratégie globale pour le patient. La synthèse des données cliniques et neuropsychométriques permet de donner une explication précise, de réaliser des aménagements de la scolarité (tiers-temps supplémentaire, élève au premier rang dans les troubles attentionnels…) et éventuellement de décider de stratégies de remédiation (orthophonie, psychomotricité…). Le médecin doit accompagner le patient et ses parents dans les démarches à l’école. Pour beaucoup de patients, il faut rédiger un protocole d’accueil individualisé (PAI) avec la conduite à tenir en cas de crise épileptique survenant à l’école. Quand il existe des difficultés scolaires, les adaptations pédagogiques à mettre en place doivent être expliquées dans le PAI. Les aménagements pédagogiques peuvent être multiples et dépendent des difficultés identifiées. Pour certains patients, un recours à la Maison départementale des personnes handicapées permet de déployer des moyens supplémentaires comme une auxiliaire de vie scolaire si cela est nécessaire ou de fournir les moyens financiers aidant à la prise en charge.
Le choix des antiépileptiques est fondé sur les données cliniques ayant démontré leur efficacité et sur la balance bénéfice-risque à l’échelle individuelle (
L’objectif du traitement est différent d’un patient à un autre. Il dépend du syndrome et de l’évolution de la maladie. Chez les enfants ayant une épilepsie débutante, l’objectif thérapeutique est d’obtenir un contrôle complet des crises épileptiques sans effet indésirable afin de permettre la vie la plus normale possible à cet enfant. En revanche, chez les enfants avec une épilepsie résistante, l’objectif n’est plus nécessairement d’être libres de crises. Chez ces patients, on cherche plutôt à améliorer la qualité de vie en évitant la survenue des crises les plus gênantes (par exemple : disparition des crises focales avec conscience altérée ou avec évolution bilatérale tonico-clonique) avec un taux d’effets indésirables le plus léger possible.
La scolarité et les apprentissages occupent une grande part de la vie des enfants. Un peu plus de 50 % des enfants atteints d’épilepsie ont des troubles scolaires. Chaque consultation doit donc comprendre une partie de dépistage de ces difficultés. Il faut s’intéresser tant aux performances qu’aux moyens mis en place pour obtenir un tel résultat. En cas de difficultés scolaires, il faut informer les parents et le médecin scolaire. Une évaluation neuropsychologique peut permettre de mieux caractériser les difficultés pour mettre en place une stratégie globale pour le patient. La synthèse des données cliniques et neuropsychométriques permet de donner une explication précise, de réaliser des aménagements de la scolarité (tiers-temps supplémentaire, élève au premier rang dans les troubles attentionnels…) et éventuellement de décider de stratégies de remédiation (orthophonie, psychomotricité…). Le médecin doit accompagner le patient et ses parents dans les démarches à l’école. Pour beaucoup de patients, il faut rédiger un protocole d’accueil individualisé (PAI) avec la conduite à tenir en cas de crise épileptique survenant à l’école. Quand il existe des difficultés scolaires, les adaptations pédagogiques à mettre en place doivent être expliquées dans le PAI. Les aménagements pédagogiques peuvent être multiples et dépendent des difficultés identifiées. Pour certains patients, un recours à la Maison départementale des personnes handicapées permet de déployer des moyens supplémentaires comme une auxiliaire de vie scolaire si cela est nécessaire ou de fournir les moyens financiers aidant à la prise en charge.
Références
1. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475-82.
2. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:512-21.
3. Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58:522-30.
4. Auvin S. Management of childhood epilepsy. Presse Med 2011;40:287-92.
5. Auvin S. Les antiépileptiques. EMC Pédiatrique. Paris: Elsevier Masson, 2013:4-091-A-013.
6. Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008;131:2264-86.
7. Garzon P, Lemelle L, Auvin S. Childhood absence epilepsy: An update. Arch Pediatr 2016;23:1176-83.
8. Beghi M, Beghi E, Cornaggia CM, Gobbi G. Idiopathic generalized epilepsies of adolescence. Epilepsia 2006;47:107-10.
2. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:512-21.
3. Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58:522-30.
4. Auvin S. Management of childhood epilepsy. Presse Med 2011;40:287-92.
5. Auvin S. Les antiépileptiques. EMC Pédiatrique. Paris: Elsevier Masson, 2013:4-091-A-013.
6. Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008;131:2264-86.
7. Garzon P, Lemelle L, Auvin S. Childhood absence epilepsy: An update. Arch Pediatr 2016;23:1176-83.
8. Beghi M, Beghi E, Cornaggia CM, Gobbi G. Idiopathic generalized epilepsies of adolescence. Epilepsia 2006;47:107-10.
Dans cet article


Résumé
Les épilepsies sont un groupe hétérogène de maladies caractérisées par la récurrence de crises épileptiques. L’incidence est plus importante chez l’enfant que chez l’adulte. Les épilepsies sont bien plus que la répétition des crises : les comorbidités sont fréquentes, de même que les restrictions dans la vie quotidienne. Chez l’enfant, il faut être particulièrement vigilant à la scolarité et aux difficultés comportementales pour mettre en place précocement une prise en charge adaptée. Les épilepsies avec crises focales sont les plus fréquentes chez l’enfant. Certains syndromes comme l’épilepsie à pointes centro-temporales, l’épilepsie-absence de l’enfant, l’épilepsie myoclonique juvénile et l’épilepsie-absence de l’adolescence représentent une majeure partie des épilepsies de l’enfant et de l’adolescent. Le diagnostic syndromique permet de réaliser des investigations appropriées, de proposer un traitement adapté et d’expliquer le pronostic.