Suspicion de fibrose pulmonaire idiopathique : diagnostiquer et surveiller
Les signes cliniques de la FPI ne sont pas spécifiques : râles crépitants secs bilatéraux (précoces et quasiment constants), dyspnée, toux non productive. Un hippocratisme digital est présent dans près de 50 % des cas. La cyanose et les signes d’insuffisance ventriculaire droite ne s’observent qu’à un stade avancé, tout comme l’amaigrissement et l’altération de l’état général.
Confirmer le diagnostic
Lorsque l’ensemble de ces signes est réuni, avec une prédominance des anomalies dans les régions sous-pleurales et dans les lobes inferieurs, on parle d’un aspect typique de pneumopathie interstitielle commune (PIC). Cet aspect est retrouvé dans 50 à 70 % des cas de FPI.
En l’absence de rayons de miel, les bronchectasies par traction associées aux réticulations permettent de parler de PIC probable (
Les explorations fonctionnelles respiratoires ne sont pas d’une grande aide pour le diagnostic, mais elles peuvent montrer :
– un trouble ventilatoire restrictif au repos caractérisé par une diminution de la capacité pulmonaire totale et de la capacité vitale forcée ;
– une diminution précoce de la capacité de diffusion du monoxyde de carbone au repos (souvent seule anomalie détectée lors du diagnostic dans les formes précoces de FPI) ;
– une hypoxémie d’abord à l’effort, puis au repos ; elle peut être facilement mise en évidence lors de tests d’exercice (test de marche de 6 minutes, test de lever de chaise) révélant une désaturation à l’effort.
Chercher une cause
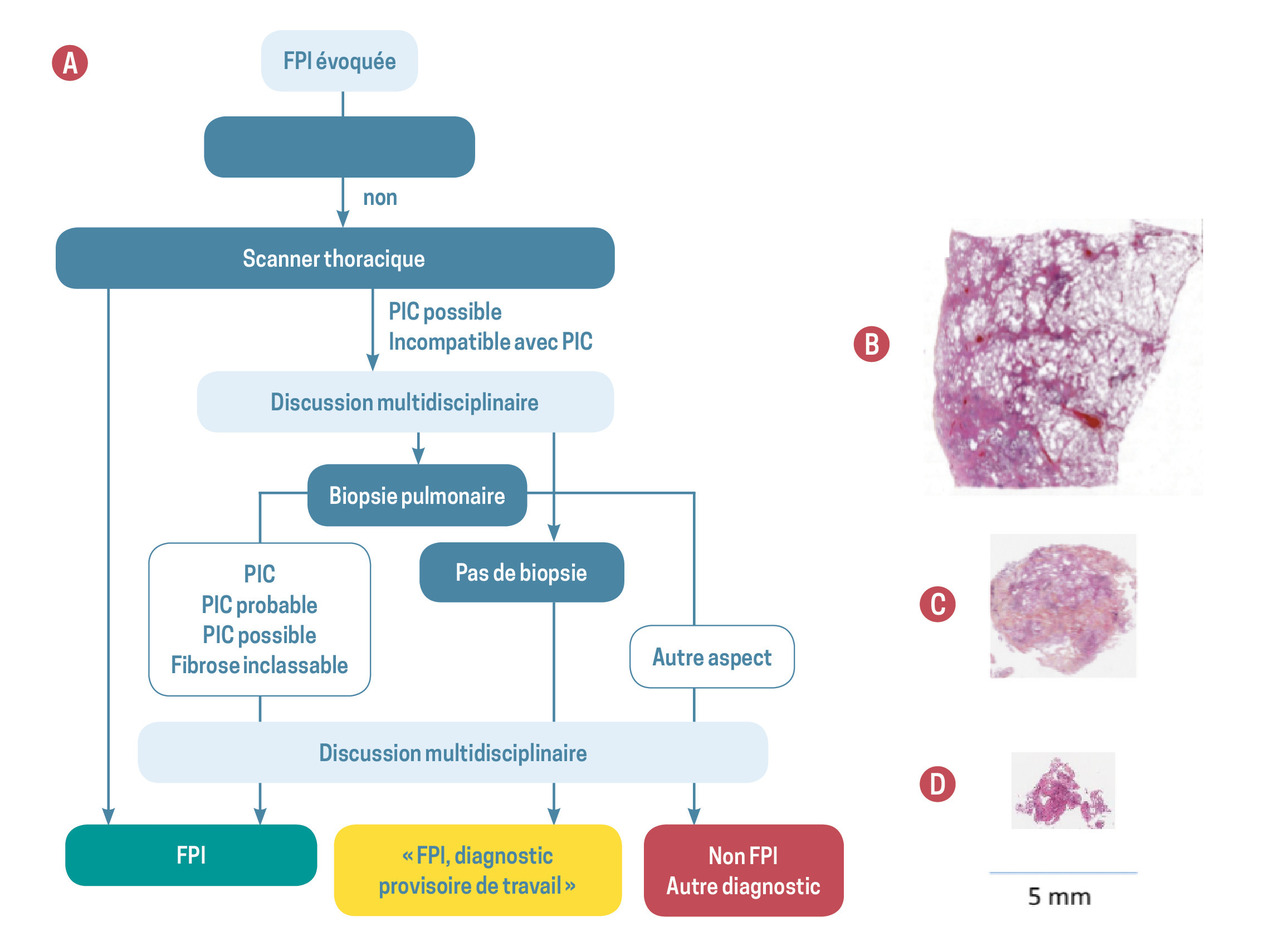
En l’absence de cause patente et lorsque l’imagerie trouve un aspect typique de pneumopathie interstitielle commune ou probable, on peut retenir le diagnostic de FPI (
Dans tous les autres cas, lorsque l’aspect du scanner n’est pas caractéristique de PIC (
La cryobiopsie transbronchique est une alternative à la biopsie pulmonaire chirurgicale, qui permet d’obtenir, au cours d’une endoscopie bronchique sous anesthésie générale, des fragments pulmonaires compatibles avec les besoins de l’analyse anatomopathologique des pneumopathies interstitielles. Il s’agit d’une technique endoscopique lors de laquelle une cryosonde est introduite par le canal opérateur. Cette sonde permet de congeler les tissus avoisinants sur plusieurs millimètres en quelques secondes. Des fragments de poumon congelé de 5 mm d’arête au moins peuvent ainsi être obtenus, ce qui permet une analyse histologique représentative du parenchyme pulmonaire (
Dans une étude prospective, si le degré de confiance dans le diagnostic était élevé, le diagnostic histologique proposé par cryobiopsie était concordant avec celui de la biopsie pulmonaire chirurgicale dans 95 % des cas. En revanche, quand le degré de confiance était plus faible, la classification était modifiée pour 23 % des patients entre cryobiopsie et biopsie chirurgicale.2,3
D’autres techniques encore moins invasives sont en cours de développement pour le diagnostic de FPI. En particulier, l’analyse transcriptomique des biopsies transbronchiques simples réalisées au cours d’une endoscopie bronchique est utilisée aux États-Unis, bien que les données soient pour l’instant encore limitées.
À noter que la recherche d’un syndrome d’apnées du sommeil associé doit également être systématique lors du diagnostic de FPI.
Évolution et surveillance
Les patients atteints de FPI sont à risque de développer une exacerbation aiguë : aggravation rapide (en moins d’un mois) de la maladie pouvant conduire le patient en réanimation, et qui doit faire éliminer une pathologie infectieuse ou thrombo-embolique spécifiquement curable. La mortalité après un tel épisode d’exacerbation est estimée à 50 %.
Le développement d’un cancer bronchique est également fréquent au cours de la FPI (5 à 10 % des patients). Ces cancers posent des problèmes spécifiques de diagnostic et de traitement : insuffisance respiratoire associée et possible exacerbation de la fibrose dans les suites des traitements oncologiques. Le dépistage de ces cancers justifie la réalisation annuelle d’un scanner thoracique.
Génétique des fibroses pulmonaires
En effet, environ 30 % des formes familiales sont expliquées par une mutation d’un gène lié à l’homéostasie des télomères, le gène TERT étant le plus fréquemment identifié. Le syndrome des télomères courts se caractérise par la présence, chez le patient ou chez un apparenté au premier degré, d’une anomalie hématologique (macrocytose, thrombopénie, voire aplasie médullaire), d’une cirrhose hépatique ou d’une canitie (blanchissement massif prématuré des cheveux, avant l’âge de 30 ans). Ces manifestations spécifiques compliquent la prise en charge et le conseil génétique des patients : après transplantation pulmonaire, ils sont plus à risque de complications hématologiques. Des adaptations des traitements immunosuppresseurs et cytotoxiques sont ainsi nécessaires.
Plus rarement, des mutations sur les gènes codant pour les protéines du surfactant sont détectées.
La susceptibilité génétique à développer une fibrose pulmonaire a été associée à un variant dans le promoteur du gène MUC5B, qui est présent dans environ 10 % de la population caucasienne. Ce variant est associé à une augmentation de la transcription de MUC5B. La présence de ce variant à l’état hétérozygote est associée à une augmentation du risque (multiplié par 6) de développer une FPI. Ce variant est détecté chez 35 % des patients atteints de FPI dans la population caucasienne. Il est beaucoup plus rare dans la population asiatique ou africaine. Il est également associé à une augmentation du risque de développer une fibrose pulmonaire non idiopathique dans des contextes étiologiques identifiés aussi variés que la polyarthrite rhumatoïde, l’asbestose ou la pneumopathie d’hypersensibilité.4 Cette découverte identifie donc des facteurs de risque génétiques commun entre la FPI et des fibroses pulmonaires secondaires, suggérant un continuum entre ces pathologies.
De plus, l’expression épithéliale de MUC5B place l’épithélium au centre de la physiopathologie de ces PID et justifie l’évaluation d’approches thérapeutiques dédiées, notamment d’études cliniques pour de nouveaux traitements par voie inhalée. Ces variants ne sont cependant pas utilisés actuellement pour le conseil génétique.
Traitement des fibroses pulmonaires
Traitements non spécifiques
Traitements de la fibrose pulmonaire idiopathique
Deux traitements antifibrosants oraux disponibles
Le nintédanib et la pirfénidone (disponibles en France depuis 2016 et 2012 respectivement) ont démontré un bénéfice dans la FPI en prise orale quotidienne.5,6Le nintédanib est un inhibiteur de la tyrosine kinase avec multicibles : VEGF (vascular endothelial growth factor), PDGF (platelet-derived growth factor), FGF (fibroblast growth factor), etc.
Le mécanisme précis de l’action de la pirfénidone est moins bien connu : probable inhibition du TGF-β (transforming growth factor beta).
Tous les patients atteints de FPI ont un déclin progressif de leur capacité vitale forcée, meilleur paramètre actuellement disponible pour évaluer l’effet d’un antifibrosant. L’efficacité des deux molécules antifibrosantes est sensiblement identique : déclin moyen de la capacité vitale forcée de 240 mL/an dans les groupes placebo contre 120 mL/an dans les groupes traités par antifibrosant. De plus, les études de cohorte ont rapporté une amélioration de la survie chez les patients ayant reçu un traitement antifibrosant.7 Ainsi, la survie moyenne actuelle après diagnostic est d’environ cinq ans.
Des effets secondaires non négligeables
Plus de 35 % des patients ont une photosensibilité secondaire à la prise de pirfénidone. Le nintédanib peut provoquer diarrhées et perte de poids. Jusqu’à 3 % des patients ont une cytolyse hépatique (transaminases augmentées à plus de 3 fois la normale) sous antifibrosants. S’il est rare de devoir arrêter le traitement pour cytolyse hépatique, plus de la moitié des patients l’arrêtent avant deux ans en raison d’une progression de la maladie ou d’une intolérance aux antifibrosants.8-10Quelles perspectives thérapeutiques ?
De nouveaux traitements antifibrosants sont à l’étude. Des résultats prometteurs ont, par exemple, été rapportés dans les essais de phase II avec le pamrevlumab (anticorps monoclonal anti-CTGF [connective tissue growth factor]).Par ailleurs, pour les patients porteurs d’une mutation sur un gène lié aux télomères, un traitement ciblé visant à activer le complexe télomérase pourrait potentiellement ralentir l’évolution de la fibrose. Ainsi, un androgène de synthèse activant le complexe télomérase fait actuellement l’objet d’un essai clinique en France, et il pourrait s’agir du premier traitement personnalisé des patients atteints de FPI.
Traitements des fibroses non idiopathiques
Certains éléments communs à certaines pneumopathies interstitielles diffuses secondaires et idiopathiques (âge, sexe, tabac, aspect scanographique de PIC et facteurs de risque génétiques) ont fait évaluer l’effet des antifibrosants dans cette indication.
Deux études cliniques récentes ont ainsi démontré que le nintédanib pouvait ralentir le déclin de la fonction respiratoire chez les patients ayant une fibrose pulmonaire dans le contexte d’une sclérodermie, ou ayant des fibroses pulmonaires progressives non idiopathiques (notamment pneumopathie d’hypersensibilité, polyarthrite rhumatoïde). En parallèle, une étude suggère que la pirfénidone ralentit l’évolution des fibroses pulmonaires progressives inclassables.11
Plusieurs autres études sont en cours pour confirmer et affiner l’intérêt de ces traitements dans les formes secondaires.
Que dire à vos patients ?
L’exposition à des toxiques inhalés (tabac, pollution, antigènes organiques par exemple) augmente le risque de développer une fibrose pulmonaire.
La fibrose pulmonaire est une maladie irréversible, mais les traitements antifibrosants en ralentissent l’évolution.
Compte tenu de la relative rareté des fibroses pulmonaires, un avis spécialisé dans un centre de référence est souvent nécessaire pour confirmer le diagnostic et orienter les thérapeutiques.
2. Ganganah O, Guo SL, Chiniah M, et al. Efficacy and safety of cryobiopsy versus forceps biopsy for interstitial lung diseases and lung tumours: A systematic review and meta-analysis. Respirol 2016;21(5):834-41.
3. Troy LK, Grainge C, Corte TJ, et al. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir Med 2020;8(2):171-81.
4. Juge PA, Lee JS, Ebstein E, et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N Engl J Med 2018;379(23):2209-19.
5. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 2011;365(12):1079-87.
6. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377(9779):1760-9.
7. Guenther A, Krauss E, Tello S, et al. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res 2018;19(1):141.
8. Cottin V, Koschel D, Günther A, et al. Long-term safety of pirfenidone: results of the prospective, observational PASSPORT study. ERJ Open Res 2018;4(4):00084-2018.
9. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med 2019;381(18):1718-27.
10. Wells AU, Flaherty KR, Brown KK, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med 2020;8(5):453-60.
11. Maher TM, Corte TJ, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomised, placebo-controlled phase II trial. BMJ Open Respir Res 2018;5(1):e000289.
12. Cottin V, Crestani B, Cadranel J, et al. Recommandations pratiques pour le diagnostic et la prise en charge de la fibrose pulmonaire idiopathique – Actualisation 2017. Version longue. Rev Mal Respir 2017;34(8):900-68.
Dans cet article
 Encadrés
Encadrés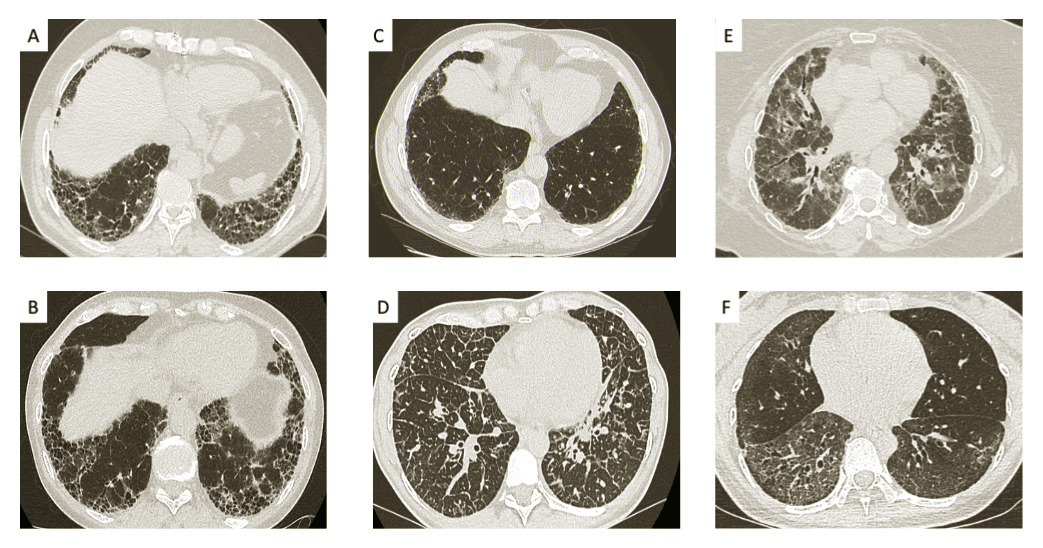

La FPI, la plus fréquente des fibroses pulmonaires, est une maladie rare, dont l’incidence annuelle est d’environ 10/100 000 personnes.
Le diagnostic repose sur l’absence de cause retrouvée et un aspect scanographique ou histologique de PIC.
La prise en charge repose sur la réhabilitation respiratoire, l’arrêt du tabac, la prévention vaccinale, un traitement antifibrosant, voire une transplantation pulmonaire.
Les fibroses pulmonaires liées à une connectivite, à une pneumopathie d’hypersensibilité ou à une exposition environnementale peuvent avoir de nombreuses similarités avec la FPI.