Une fièvre récurrente chez l’enfant doit faire évoquer une maladie auto-inflammatoire (MAI), secondaire à l’activation excessive du système immunitaire inné médiée par des cytokines, notamment l’interleukine 1 (IL-1). La plus fréquente est le PFAPA (Periodic Fever, Aphthous stomatitis, Pharyngitis, cervical Adenitis), devant les affections récurrentes héréditaires telles que la fièvre méditerranéenne familiale (FMF) et les cryopyrinopathies. Ces dernières sont constituées de 3 phénotypes de gravité croissante : urticaire familiale au froid (FCAS), syndrome de Muckle-Wells (MWS) et syndrome chronique inflammatoire cutané et articulaire (CINCA). Plus rares : déficit en mévalonate kinase (MKD) et syndrome TRAPS (TNF receptor associated periodic syndrome ;
Quand l’évoquer ?
Elle est définie par des épisodes fébriles récidivants (≥ 3 épisodes) nus ou associés à une symptomatologie stéréotypée, limitée dans le temps, entrecoupés d’intervalles libres apyrétiques se reproduisant pendant des mois ou des années.
Ces fièvres peuvent être dites périodiques ou non, selon le rythme de survenue des crises. Elles sont à distinguer des fièvres récidivantes ou intermittentes caractérisées par des épisodes répétés mais non stéréotypés et sans périodicité, et des hyperthermies continues qui appartiennent à un autre champ nosologique.
Cliniquement, les épisodes récurrents débutent généralement dans l’enfance, voire en période néonatale, et peuvent être accompagnés d’éruptions cutanées, de sérites, d’arthromyalgies, d’une atteinte digestive, ophtalmologique ou neurosensorielle dans les formes sévères. Au cours d’un accès de MAI, on ne retrouve pas de signes ORL de type rhinite ou toux ni de réponse partielle aux antipyrétiques. Un syndrome inflammatoire biologique (CRP) avec polynucléose est systématiquement décrit. Tout comme la symptomatologie clinique, ce dernier diminue, voire régresse spontanément en dehors des poussées.
Un facteur déclenchant est parfois identifié : stress environnemental, fatigue, exposition au froid dans les CAPS, épisodes infectieux ou vaccination pour le MKD. La fréquence et la durée des épisodes inflammatoires sont variables en fonction de la MAI (
On peut diagnostiquer des formes atypiques et/ou peu sévères à l’âge adulte, les premières crises ayant été assimilées à des épisodes viraux infantiles.
Démarche diagnostique
L’interrogatoire, étape essentielle, met en évidence le caractère stéréotypé des épisodes et l’absence de symptômes entre les crises. Il faut préciser leur durée, les symptômes associés, les facteurs déclenchants, les antécédents familiaux, l’origine ethnique (arbre généalogique en remontant jusqu’aux grands-parents ou arrière-grands-parents) et l’âge de début qui varient selon le type de MAI (
L’examen clinique objective les symptômes présents pendant et en dehors des crises fébriles. La courbe de croissance est à surveiller. En effet, une mauvaise croissance staturo-pondérale – qui certes peut compliquer certaines MAI rares et sévères – doit faire évoquer un diagnostic différentiel.
Objectifs des explorations biologiques : rechercher un syndrome inflammatoire avec une hyperleucocytose à polynucléaires neutrophiles durant la crise (NFS, CRP) s’améliorant et/ou disparaissant en dehors. Éliminer les diagnostics différentiels : infections (lors de la crise), déficit immunitaire, neutropénie cyclique, néoplasie et/ou maladie inflammatoire chronique (bilan hors crise).
PFAPA : la plus fréquente
Le PFAPA (fièvre périodique, stomatite aphteuse, pharyngite, adénopathie) est la plus courante des MAI non héréditaires avant 5 ans, son incidence étant évaluée à 2-3 pour 10 000 d’après une étude norvégienne.2
Les symptômes débutent le plus souvent dans la première année de vie et surviennent à intervalles réguliers de 3 à 6 semaines, ce qui les distingue des autres MAI. Durée médiane de chaque épisode : 3 à 6 jours. Il se manifeste par une fièvre élevée (40 °C, parfois mal tolérée) avec au moins un des principaux symptômes : pharyngite, adénite cervicale ou aphtose buccale.3 Surviennent parfois des douleurs abdominales péri-ombilicales, des arthromyalgies sans signes d’arthrite et des céphalées lors des crises. En dehors, bon état général et croissance staturo- pondéral parfaite.
Le syndrome inflammatoire marqué lors des crises (CRP > 50 mg/L en moyenne) se normalise dans l’intervalle. Le PFAPA est le plus souvent spontanément résolutif dans l’enfance, vers l’âge de 6 ans (ou une médiane de 4 ans d’évolution).
Diagnostic différentiel : le déficit en mévalonate kinase. Il est important de doser l’acidurie mévalonique lors d’une crise, qui sera alors augmenté en cas de MKD.
Le traitement est surtout symptomatique : AINS (ibuprofène à la dose de 20 à 30 mg/kg/j) ou les glucocorticoïdes en une prise unique dans les 12 premières heures de la crise : 1 mg/kg de prednisone (au maximum 60 mg) ou 0,1 mg/kg de prednisolone ou 10 gouttes/kg de bêtaméthasone. Non efficace, l’amygdalectomie n’est plus recommandée. Elle peut être proposée dans certaines indications ORL : apnées du sommeil, angines bactériennes récurrentes.
FMF : pas de périodicité
MAI autosomique récessive, elle doit être évoquée chez les patients d’origine méditerranéenne (populations arabes, arméniennes, séfarades, turques, libanaises, italiennes et grecques). Elle débute le plus souvent avant 20 ans (4 ans en moyenne) et se caractérise par des épisodes récurrents de fièvre (38,5-40 °C) de courte durée (1-3 jours), classiquement associée à un malaise général et des frissons. Contrairement au PFAPA, pas de périodicité (fréquence allant de 1 x/semaine à 1 x/an). Entre, les patients sont le plus souvent asymptomatiques.
Lors des crises, l’inflammation des séreuses entraîne une atteinte péritonéale (95 %), articulaire (mono-arthrite 75 %), pleurale (39 %), péricardique (2 %) et testiculaire (9 %) chez les hommes. L’atteinte cutanée, plus aléatoire (7-40 %), se manifeste classiquement par un pseudo- érysipèle des membres inférieurs, souvent pris pour une cellulite infectieuse ou une phlébite.4
Le diagnostic est essentiellement clinique,5 confirmé par l’étude du gène MEFV. Sur le plan biologique, aucun marqueur n’est spécifique. Le syndrome inflammatoire en période de crise (CRP, SAA ou protéine amyloïde sérique A) régresse dans la plupart des cas en dehors.
Traitement : essentiellement colchicine au long cours (chez l’enfant, la dose initiale est de 0,5 mg/j avant 5 ans, de 0,75 mg/j entre 5 à 10 ans et de 1 mg/j à 10 ans et plus) avec AINS et/ou antalgique lors des épisodes fébriles. La dose de colchicine ne doit pas être augmentée lors des fièvres sous peine d’intoxication. Elle a pour but d’éviter l’amylose rénale (par dépôt de complexes macroprotéiques, dont la SAA, se localisant principalement dans les reins et la muqueuse digestive). L’accroissement des doses dépend de la réponse initiale évaluée après au moins 3 mois d’évolution. Les patients dits « non répondeurs » à la colchicine sont très rares. La première cause est la non- observance à la colchicine. En cas de réponse insuffisante, un avis en centre de référence est indiqué pour discuter la mise en place d’un anti-IL-1.
Pronostic : plutôt bon
Cependant, les épisodes récurrents peuvent impacter la qualité de vie, les relations socio-affectives et familiales, et entraîner un absentéisme scolaire important.
L’amylose secondaire de type AA, complication tardive classique, impose une prévention par un suivi régulier et un traitement efficace de l’inflammation biologique (cible : CRP < 5 mg/L chez l’enfant ou SAA < 9 mg/L) ; sa fréquence de survenue dépend du type de maladie et de son phénotype.
Le traitement, variable selon les MAI, a pour objectif de réduire l’activité de la maladie, en particulier le nombre et l’intensité des crises ; d’améliorer la qualité de vie et de prévenir les complications dominées par l’amylose AA dans les pathologies médiées par l’IL-1.
Qui tester sur le plan génétique ?
Ils sont inutiles en cas de PFAPA. Si l’évolution est inhabituelle, il est préférable d’adresser l’enfant à un centre de référence, l’analyse génétique n’étant souvent pas contributive. Chez des patients d’origine méditerranéenne ayant les critères cliniques, on peut demander la recherche de mutation du gène MEFV. Deux mutations dans l’exon 10 affirment le diagnostic. En cas d’hétérozygotie, il est préférable de demander l’avis du centre de référence. En effet, certains patients peuvent être symptomatiques et requièrent un traitement par colchicine. Pour les autres, l’orientation clinique doit guider les recherches génétiques. Certains variants de séquence retrouvés dans la population générale peuvent induire une mésinterprétation. Un avis d’experts est précieux, d’autant que beaucoup de MAI restent à ce jour encore inclassées.
1. Piram M, Koné-Paut I, Lachmann HJ, et al. Validation of the auto-inflammatory diseases activity index (AIDAI) for hereditary recurrent fever syndromes. Ann Rheum Dis 2014;73:2168-73.
2. Førsvoll J, Kristoffersen EK, Øymar K. Incidence, clinical characteristics and outcome in Norwegian children with periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome; a population-based study. Acta Paediatr 2013;102:187‑92.
3. Gattorno M, Hofer M, Federici S, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis 2019;78:1025‑32.
4. Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol 2013;147:155‑74.
5. Yalçınkaya F, Özen S, Özçakar ZB, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford) 2009;48:395‑8.
Dans cet article
 Encadrés
Encadrés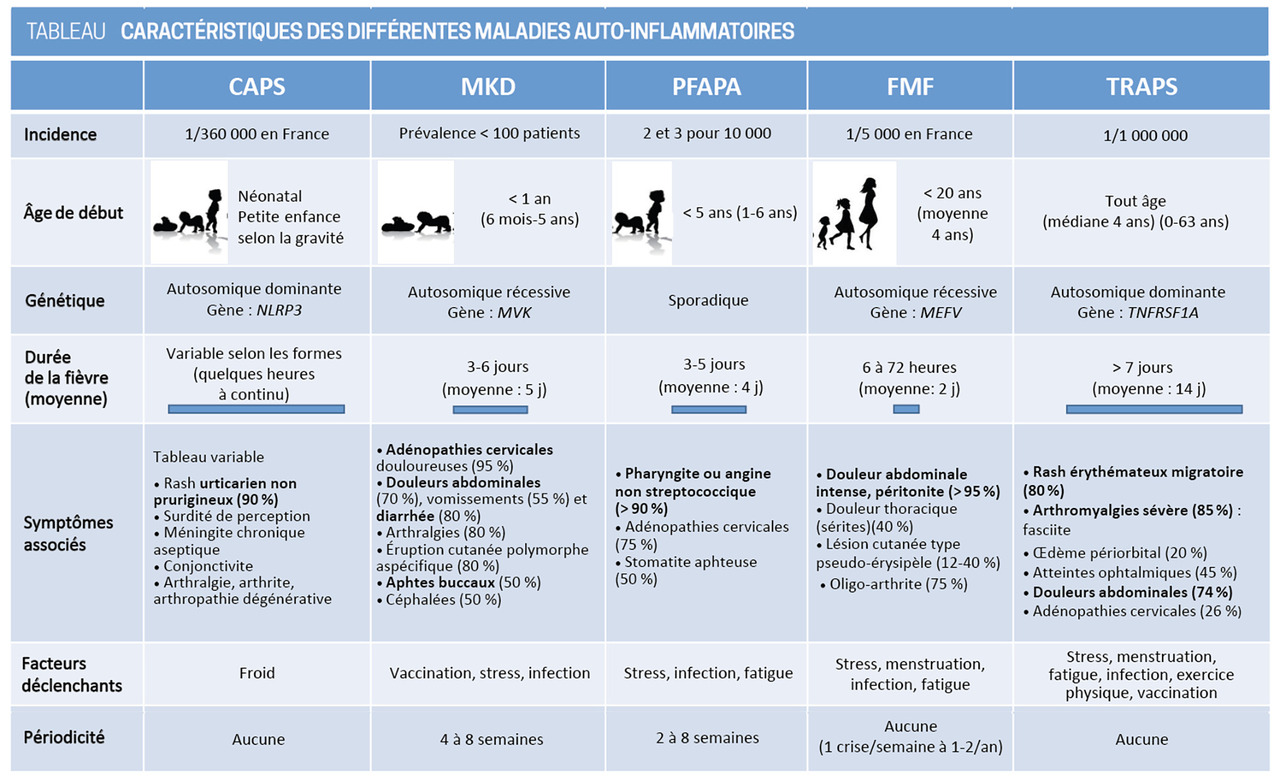

Évoquer une MAI devant une fièvre récurrente stéréotypée avec syndrome inflammatoire en l’absence d’autres causes (infections, néoplasie…).
Durée, périodicité, âge de début, symptômes associés et origine ethnique orientent vers une cause.
Le PFAPA (fièvre périodique, stomatite aphteuse, pharyngite, adénopathie) est la plus fréquente avant 5 ans.
Prise en charge spécialisée pour éviter la redoutable amylose secondaire de type AA.