La sclérose en plaques (SEP) est une maladie neuro-inflammatoire chronique du système nerveux central (SNC). Elle est caractérisée par la présence de lésions (ou plaques) au niveau du cerveau et/ou de la moelle épinière et/ou des nerfs optiques. Au sein de ces lésions inflammatoires, la gaine de myéline entourant les axones des neurones est partiellement ou totalement détruite. Or la myéline sert à isoler et protéger les prolongements neuronaux, participant à l’accélération de la conduction nerveuse. Lorsqu’il se chronicise, le phénomène de démyélinisation entraîne une dégénérescence axonale conduisant à une perte neuronale et une diminution du volume cérébral.
Une origine plurifactorielle
La cause exacte de la sclérose en plaques reste encore mal connue. Elle est considérée comme d’origine multifactorielle, impliquant à la fois des facteurs génétiques et environnementaux.
Susceptibilité génétique : de nombreux variants alléliques impliqués
La sclérose en plaques n’est pas une maladie génétique à transmission mendélienne. Cependant, des études épidémiologiques familiales ont montré l’implication de la génétique : le risque de développer une SEP est de 30 % chez une personne dont le jumeau monozygote est lui-même atteint ; cette prédisposition diminue jusqu’à 5 % pour les jumeaux dizygotes. De plus, de vastes études d’associations pangénomiques* ont permis d’identifier plus de 230 variants alléliques associés à la SEP.1 Par exemple, l’allèle HLA-DRB*15:01 confère un risque plus élevé de développer la maladie ; d’autres allèles, tel que HLA*02:01, sont au contraire plutôt protecteurs de la maladie. Un patient atteint de SEP peut présenter plusieurs de ces variants, tout comme il peut n’en présenter aucun. Si les gènes de susceptibilité de la SEP ne suffisent donc pas à expliquer la survenue de la maladie, il est intéressant de noter que la grande majorité de ces variants sont liés au système immunitaire.
Des facteurs environnementaux dominés par l’infection à EBV
Le contributeur environnemental le plus notable est probablement l’infection au virus d’Epstein-Barr (EBV). Ce virus infecte certes environ 95 % de la population adulte mais la séropositivité des patients atteints de sclérose en plaques s’élève à 99 %. Une étude récente sur une cohorte de 10 millions de jeunes adultes suivie pendant vingt ans révèle qu’une infection par l’EBV augmenterait le risque de développer la maladie jusqu’à 32 fois.2
Un lien géographique est également établi avec un gradient de latitude nord-sud dans l’hémisphère Nord et un gradient sud-nord dans l’hémisphère Sud. L’Amérique du Nord et l’Europe sont ainsi considérées comme des zones à forte prévalence de la maladie. Le gradient nord-sud est même retrouvé à l’échelle nationale de la France.
Le manque d’exposition solaire est aussi noté, corrélé à une déficience en vitamine D.
L’obésité infantile (de l’enfant au jeune adulte) doublerait le risque de d’être atteint d’une sclérose en plaques.
La consommation de tabac est non seulement associée à un risque accru de développer la maladie mais également à une augmentation du taux de transition vers une forme progressive.3
Enfin, le dérèglement du rythme circadien est un autre facteur de risque identifié de sclérose en plaques.
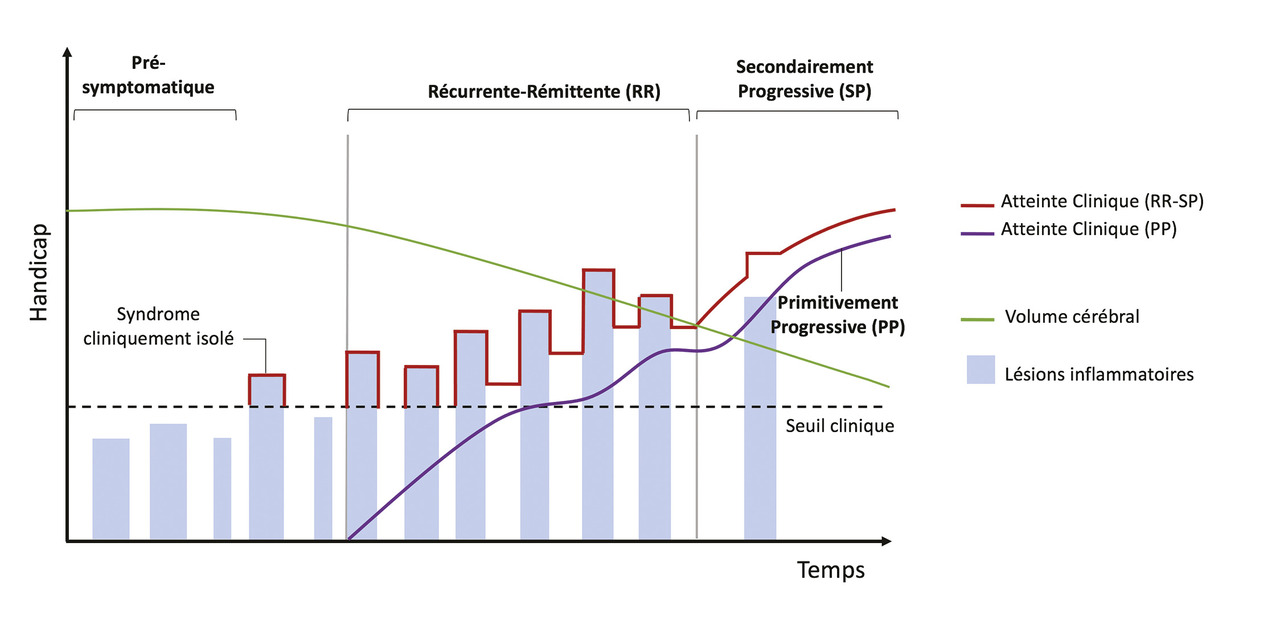
Évolution et activité : deux paramètres pour classer la maladie
La maladie débute entre 20 et 40 ans pour la majorité des patients, mais peut également concerner des personnes plus jeunes ou plus âgées. Lorsque les patients subissent une poussée, de nouveaux symptômes apparaissent (variables selon les zones du système nerveux central touchées par les lésions) ou des symptômes préexistants s’aggravent. Ces poussées peuvent durer de quelques jours à plusieurs semaines.
Évolution de la maladie : quatre formes cliniques
Classiquement, on distingue quatre formes cliniques de sclérose en plaques selon son évolution.4
Dans 85 % des cas, les patients présentent une forme de la maladie dite « récurrente-rémittente » (RR), caractérisée par l’apparition de symptômes sous forme de poussées suivies de périodes de rémission plus ou moins longues.
Après quinze à vingt ans de maladie en moyenne, en l’absence de traitement, la plupart des patients basculent vers une forme de progression lente sans rémission, dite « secondairement progressive » (SP).
Chez 8 à 10 % des patients, en particulier ceux ayant débuté la maladie à un âge plus avancé, la SEP se présente sous forme « primaire progressive » (PP), avec une accumulation de handicaps dès le début de la maladie (
Enfin, la forme « progressive récurrente » (PR) est la moins fréquente (moins de 5 % des patients). Elle ressemble à la forme « primaire progressive » à laquelle s’ajoute l’apparition de poussées récurrentes.
La SEP rémittente récurrente est associée à l’apparition de nouvelles lésions visibles en imagerie par résonance magnétique (IRM) ; la SEP progressive (primaire ou secondaire) est plutôt associée à l’augmentation graduelle de l’atrophie cérébrale et médullaire et plus rarement à l’apparition de nouvelles lésions de la substance blanche visibles en IRM. La récurrence des poussées, la présence de séquelles et la vitesse de progression de la maladie sont des paramètres très hétérogènes selon les individus. Cependant, les formes PP et SP montrent une vitesse de progression similaire, indépendante des précédentes poussées inflammatoires.
Nouvelle classification plus orientée vers la pratique
Cette classification en quatre formes cliniques a fait l’objet d’une récente mise à jour, fondée sur la notion d’« activité » de la maladie, plus en accord avec sa prise en charge pratique. En effet, les formes « actives » et « non actives » de la SEP peuvent être différenciées, qu’elle soit récurrente ou progressive. Les formes « actives » sont déterminées par des poussées récurrentes et/ou la détection de nouvelles lésions T2 à l’IRM. La SEP peut ainsi être à la fois progressive et active.
La SEP : une maladie auto-immune ?
De nombreux arguments plaident pour une origine auto-immune de la sclérose en plaques. La majorité des variants génétiques associés à la SEP sont liés au système immunitaire : système HLA (human leukocyte antigen), récepteurs aux interleukines 2 et 7, récepteurs du TNF (tumor necrosis factor), etc.
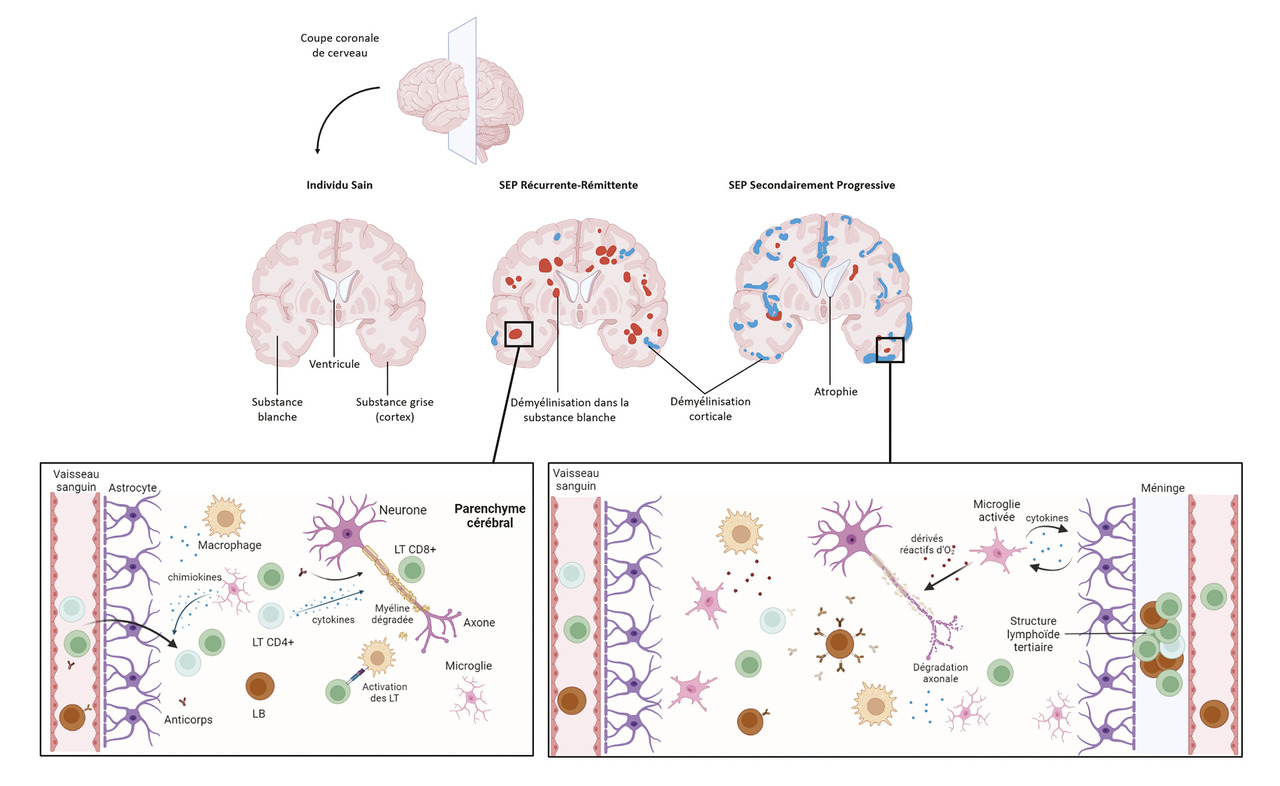
Le système nerveux central, en tant qu’organe immunoprivilégié, est protégé par des barrières telle la barrière hémato-encéphalique (BHE). Malgré cela, chez des patients atteints de SEP, le système nerveux central (SNC) subit une inflammation induite par l’activation de cellules immunitaires résidentes ainsi que par l’intrusion de cellules immunitaires périphériques passant à travers la BHE.
Les traitements actuels de la SEP sont immunomodulateurs ou immunosuppresseurs. Ils ciblent les cellules immunitaires soit de façon générale (la mitoxantrone inhibe la synthèse et la réparation de l’ADN), soit de manière plus ciblée par des anticorps monoclonaux (déplétion de lymphocytes B par anticorps anti-CD20) ou par des molécules bloquant la migration des leucocytes (par exemple, blocage de la molécule d’adhésion VLA-4 par le natalizumab).
Immunopathologie : des mécanismes cellulaires multiples, intriqués et évolutifs
Les principales cellules de l’immunité innée retrouvées dans les lésions de sclérose en plaques sont les macrophages et les microglies. Les macrophages font partie des cellules périphériques infiltrant les lésions en grand nombre, avec un phénotype pro-inflammatoire. Les microglies sont des cellules résidentes du SNC, impliquées dans la surveillance immune, capables de présenter des antigènes et de phagocyter, comme les macrophages.
Activés en réponse à des stimuli, ces deux types de cellules produisent des cytokines (contribuant à l’environnement inflammatoire) et des chimiokines (pour attirer et stimuler les cellules de l’immunité adaptative comme les lymphocytes). De plus, les microglies peuvent activer les astrocytes résidant également dans le SNC qui, à leur tour, recrutent et activent davantage de microglies et inhibent la remyélinisation en empêchant la maturation des oligodendrocytes formant la myéline.
Les lymphocytes T sont des acteurs essentiels
La majorité des lymphocytes retrouvés au sein des lésions sont les lymphocytes T (LT), en majorité de type CD8+ (cytotoxique), même si des lymphocytes T CD4+ (helper) sont aussi présents.5 La démyélinisation étant un aspect fondamental de la maladie, il est fortement suspecté que des antigènes dérivés des protéines de la myéline sont ciblés par des LT autoréactifs. Cependant, à ce jour, la communauté scientifique n’a pas encore mis en évidence un auto-antigène spécifique de la SEP reconnu par les LT CD8+.
Les LT CD8+ présents dans le SNC sont des cellules cytotoxiques bien armées pour induire directement des lésions. Une fois infiltrées, elles acquièrent un phénotype mémoire et deviennent résidentes dans le SNC.
Les LT CD4+ sont également retrouvés dans les lésions, bien qu’en quantité moindre. Ils sont essentiellement présents sous leurs formes différenciées T helper 1 (Th1) et T helper 17 (Th17), deux phénotypes pro-inflammatoires produisant des cytokines telles que les interleukines 2 et 17 et l’interféron gamma.6 Les LT CD4+ semblent surtout jouer un rôle dans l’initiation de la réponse immune, notamment en périphérie et au contact des lymphocytes B. Leur rôle est moindre dans les stades suivants car ils sont peu présents, voire absents, du tissu lors de l’apparition de nouvelles lésions.
Des travaux récents ont montré que les lymphocytes T CD4+ présents dans les lésions pouvaient reconnaître certains antigènes neuronaux. En situation normale, les LT autoréactifs sont sous le contrôle d’autres lymphocytes régulateurs (Tregs). Il est possible que la fonction suppressive de ces cellules soit altérée dans la SEP car ils ne parviennent pas à réduire suffisamment le nombre des LT cytotoxiques, surtout aux stades précoces de la maladie.
Lymphocytes B : pas en reste !
Le rôle des lymphocytes B (LB) dans la physiopathologie de la sclérose en plaques a longtemps été sous-estimé par rapport à celui des lymphocytes T. Or de plus en plus d’études démontrent leur importance.
Ces cellules sont bien présentes dans le SNC, que ce soit dans le parenchyme, les méninges ou le liquide céphalorachidien (LCR) : plus de 95 % des patients ont une réponse humorale intrathécale, avec une sécrétion d’immunoglobulines de type G (IgG), et parfois IgM, qui peuvent être détectées dans le LCR.
Les LB peuvent également sécréter des cytokines et activer les LT par leur rôle de cellules présentatrices d’antigène. Il semble d’ailleurs que cette dernière fonction soit primordiale dans les mécanismes physiopathologiques conduisant à la SEP : dans le sang périphérique des patients, la présence de LB autoproliférants a été mise en évidence ; ces cellules contrôlent la prolifération des LT, notamment CD4+ grâce à la présentation d’antigène. La déplétion des LB par des anticorps monoclonaux anti-CD20 tels que le rituximab, l’ocrélizumab ou l’ofatumumab s’est avérée efficace pour prévenir la survenue de poussées cliniques et de nouvelles lésions en IRM, démontrant ainsi leur importance dans l’immunopathologie de la SEP.7
Des structures méningées lymphoïdes tertiaires
Lorsque les méninges sont inflammées, des lymphocytes T et B peuvent y apparaître sous forme diffuse, mais aussi sous forme de structures lymphoïdes tertiaires avec des agrégats denses et compacts dans les cas les plus sévères.8 Ces structures sont détectées après de nombreuses années d’inflammation chronique chez les patients atteints de forme secondairement progressive (SP), ainsi que chez les patients atteints de forme primaire progressive (PP) avec une évolution rapide de la sclérose en plaques.
Profil cellulaire évoluant avec la maladie
L’inflammation est présente à tous les stades de la SEP, mais elle est plus prononcée lors des phases aiguës de poussée que lors des phases de progression. Les premières lésions sont fortement caractérisées par une infiltration de cellules immunitaires périphériques à travers la barrière hémato-encéphalique, que l’on retrouve notamment au niveau périvasculaire. Les plus nombreuses sont les macrophages, suivies des lymphocytes T CD8+, puis des LT CD4+ ; les lymphocytes B sont moins nombreux et représentent environ 5 % des cellules infiltrantes.
Lorsque la maladie progresse, les infiltrats B et T sont plus diffus, et la proportion de lymphocytes B augmente. Les macrophages résidents restent activés. On note également une très forte présence de microglies activées, qui peuvent sécréter des médiateurs neurotoxiques tels que les dérivés réactifs de l’oxygène.9 La démyélinisation est plus diffuse, et la dégénérescence axonale devient évidente.
Neuropathologie
La sclérose en plaques se distingue des autres maladies du SNC par la présence de lésions focales dans les substances blanches et grises. Le tissu endommagé par ces lésions de démyélinisation peut entrer en phase de remyélinisation partielle ou complète, le degré de remyélinisation variant fortement d’un patient à l’autre.
Lors des stades précoces de la maladie, des lésions actives apparaissent surtout dans la substance blanche, accompagnées d’une atteinte de la barrière hémato-encéphalique. Malgré les infiltrats immunitaires, et hormis les zones contenant les lésions, le SNC reste relativement peu détérioré : la substance blanche est dite « d’apparence normale ». Ces lésions apparaissent ensuite plus rarement dans la substance blanche chez les patients évoluant vers des formes progressives.
Avec l’âge et au fil du temps, les lésions actives ou chroniques actives préexistantes connaissent une lente expansion. Les lésions de la substance grise responsables de la démyélinisation corticale deviennent alors plus nombreuses, possiblement en relation avec les infiltrats méningés en regard, de même que les atteintes de substance blanche plus diffuses (
Des mécanismes physiopathologiques de mieux en mieux identifiés
La sclérose en plaques débute avec des lésions profondes de la substance blanche ayant des caractères inflammatoires focaux aux stades précoces (récurrente-rémittente), surtout dépendants de l’infiltration depuis la périphérie de lymphocytes T et de macrophages. Ceux-ci laissent place à une atteinte inflammatoire diffuse aux stades plus tardifs (secondairement progressive) surtout dépendante d’une activation microgliale et macrophagique.
* NDLR : une étude d’association pangénomique compare les données génétiques de milliers, voire de millions, d’individus pour trouver les polymorphismes mononucléotidiques particuliers qui sont associés au risque de développer une maladie particulière ou d’avoir un trait particulier.
1. International Multiple Sclerosis Genetics Consortium. Low-frequency and rare-coding variation contributes to multiple sclerosis risk. Cell 2018;175(6):1679-87.e7.
2. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022 21;375(6578): 296-301.
3. Ascherio A, Munger K. Epidemiology of Multiple sclerosis: from risk factors to prevention - An update. Semin Neurol 2016 26;36(02):103-14.
4. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS. Defining the clinical course of multiple sclerosis. Neurology 2014;9.
5. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. Journal of the Neurological Sciences 1983;62(1-3):219-32.
6. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol 2015;15(9):545-58.
7. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376:209-20.
8. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathology 2004;14(2):164-74.
9. Voet S, Prinz M, van Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends in Molecular Medicine 2019;25(2):112-23.
10. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol 2019;9:3116.