Toujours y penser devant un syndrome œdémateux, en raison des complications immédiates potentiellement graves et du risque d’évolution vers l’insuffisance rénale terminale. De nombreuses causes doivent être recherchées rapidement.
La glomérulonéphrite extramembraneuse est la principale cause de syndrome néphrotique de l’adulte parmi les maladies glomérulaires primitives. Elle survient préférentiellement entre 50 et 60 ans avec une prédominance masculine (2 hommes pour 1 femme).1 Son incidence est estimée à 10 à 20 personnes par million par an.1 En dépit de cette relative rareté, ce diagnostic doit être systématiquement envisagé devant un syndrome œdémateux, en raison des éventuelles causes à identifier, des complications immédiates potentiellement graves et/ou du risque d’évolution vers l’insuffisance rénale terminale.
Si la grande majorité des glomérulonéphrites extramembraneuses survient isolément, définissant la forme dite primaire, (anciennement appelée « idiopathique »), 20 % des glomérulonéphrites extramembraneuses sont dites secondaires à une cause établie ou présumée (
Physiopathologie
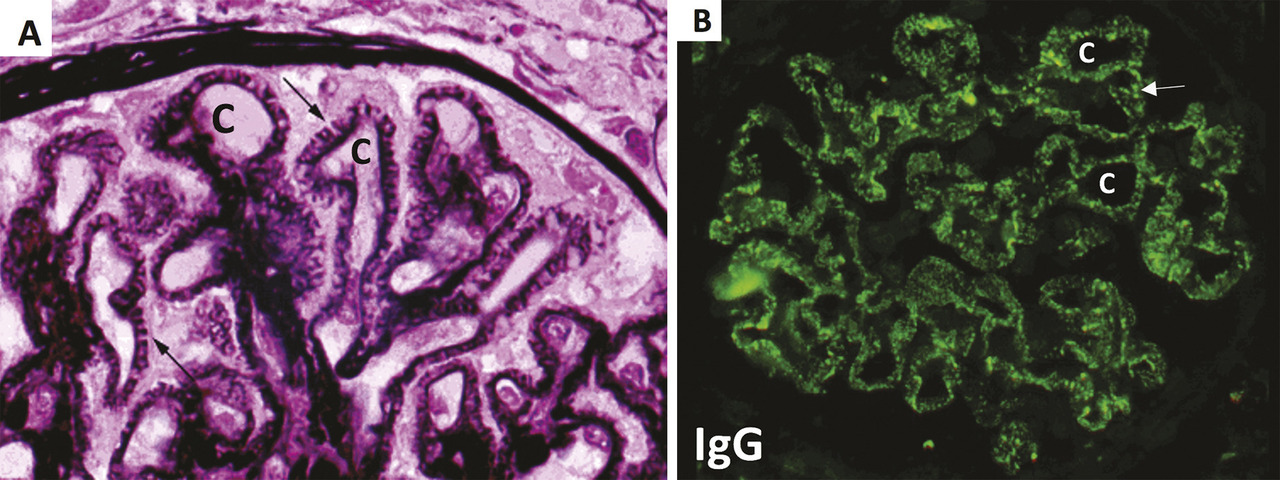
Le dépôt sur le versant externe podocytaire (sous-épithélial) de la membrane basale glomérulaire de complexes immuns constitués d’immunoglobulines G (IgG) et de fractions du complément constitue la lésion élémentaire de la glomérulonéphrite extramembraneuse. L’activation locale du complément qui aboutit à la formation du complexe d’attaque membranaire C5b9 est responsable d’altérations fonctionnelles et cytosquelettiques modifiant la microarchitecture podocytaire. Il en résulte une altération du filtre glomérulaire responsable d’un passage massif de l’albumine sérique dans les urines définissant le syndrome néphrotique, lui-même responsable de complications (insuffisance rénale fonctionnelle, dyslipidémie, hypercoagulabilité, hypogammaglobulinémie, dénutrition).
Découverte des antigènes cibles podocytaires
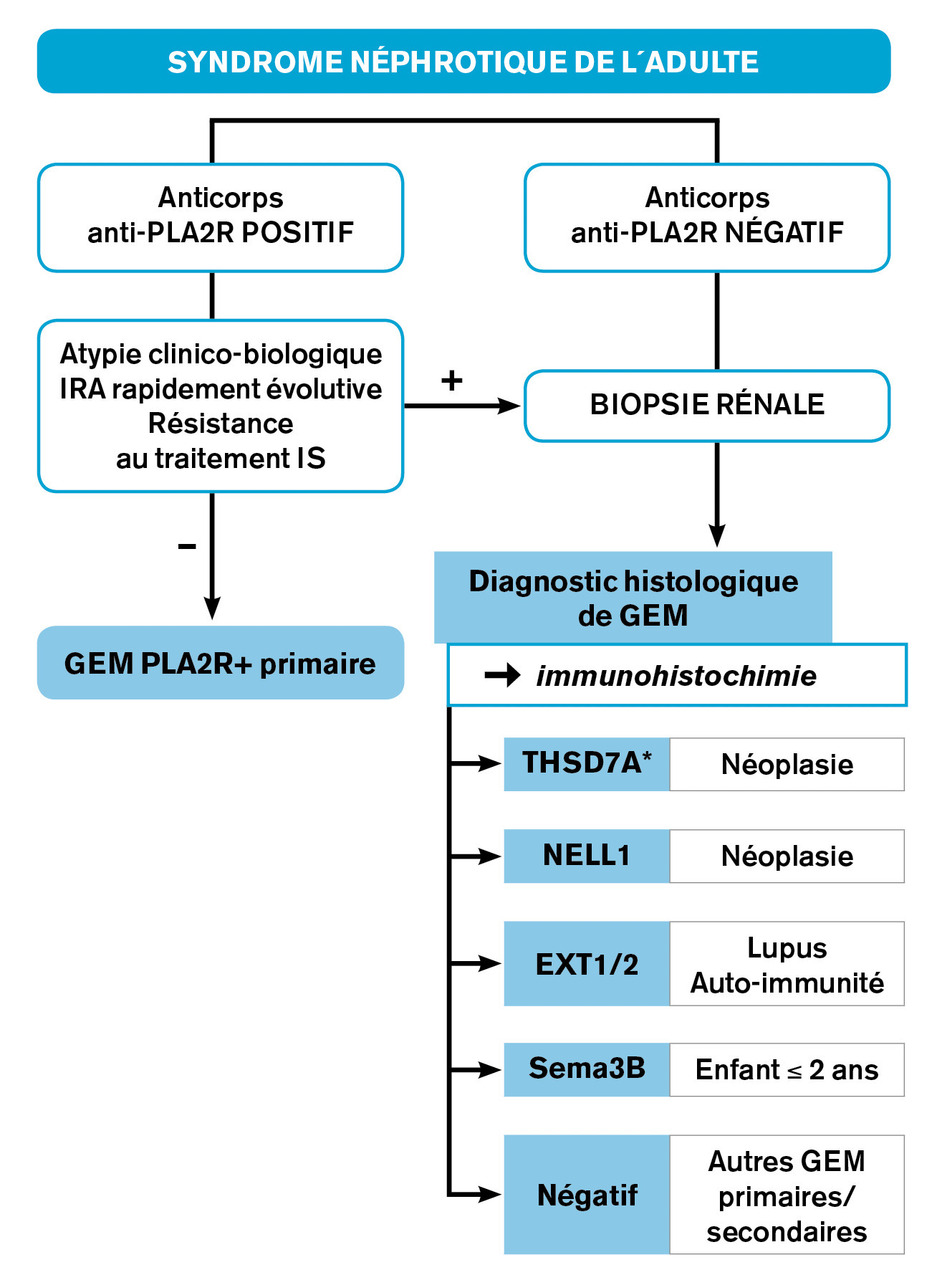
L’origine auto-immune des formes primaires de glomérulonéphrite extramembraneuse a été suggérée par l’étude de modèles expérimentaux chez le rat (néphrite de Heymann), permettant, dans les années 1980, l’identification d’auto-anticorps dirigés contre la mégaline/LRP2 – antigène exprimé par le podocyte dans cette espèce, mais dont l’expression est restreinte au tube proximal chez l’homme.2 L’endopeptidase neutre (EPN) est le premier antigène podocytaire identifié chez l’homme dans des formes exceptionnelles de glomérulonéphrites extramembraneuses néonatales, résultant d’une allo-immunisation materno-fœtale induite par un déficit génétique complet en EPN chez la mère.3 Dans le sillage de cette découverte, l’identification en 2009 du récepteur de la phospholipase A2 (PLA2R) comme auto-antigène cible dans environ 70 % des cas de glomérulonéphrites extramembraneuses primaires de l’adulte a constitué une avancée physiopathologique majeure.2, 3 Depuis, de nouveaux antigènes ont été caractérisés, dont la fréquence cumulative est estimée entre 15 et 20 % des cas de glomérulonéphrite extramembraneuse. Il s’agit de la thrombospondine de type 1 contenant un domaine 7A (THSD7A), de la protéine analogue au facteur de croissance épidermique neural 1 (NELL-1), des exostosines 1 et 2 (EXT1/EXT2), et de la sémaphorine 3B (Sema3B) [
Pour tous ces antigènes, à l’exception des EXT1/EXT2, des auto-anticorps spécifiques ont pu être mis en évidence dans le sérum des patients. Les anticorps anti-PLA2R, désormais largement utilisés en pratique clinique, constituent un biomarqueur performant grâce à leur spécificité proche de 100 % et leur valeur prédictive pour l’évolution de la maladie et la réponse au traitement immunosuppresseur.6, 7 En l’absence d’insuffisance rénale, d’atypies cliniques et/ou signes extrarénaux, la détection des anticorps PLA2R permet de surseoir à la biopsie rénale pour établir le diagnostic de glomérulonéphrite extramembraneuse. En outre, les taux d’anticorps anti-PLA2R sont corrélés avec l’activité de la maladie : un titre faible au diagnostic est prédictif de rémission spontanée, incitant à un traitement conservateur ; à l’inverse, un taux basal élevé ou son augmentation sont associés à la persistance du syndrome néphrotiqueet à un risque accru de déclin fonctionnel rénal, posant l’indication d’un traitement immunosuppresseur. Au cours de l’évolution, la négativation du taux des anti-PLA2R est prédictive de rémission clinique, tandis que la réapparition ou l’augmentation du titre d’anticorps précède la rechute clinique. Enfin, leur persistance ou leur réapparition après une greffe rénale expose à un risque accru de récidive sur le greffon.
Bases génétiques
Les avancées récentes de la génétique ont permis de démontrer l’existence de facteurs génétiques de prédisposition à la survenue d’une glomérulonéphrite extramembraneuse.8 Une étude pangénomique (GWAS) réunissant 556 patients caucasiens européens a permis d’identifier des allèles de prédisposition sur deux locus distincts, associés de façon très significative avec la glomérulonéphrite extramembraneuse primaire : le premier dans le gène PLA2R1 (locus 2q24) codant l’antigène cible majeur PLA2R ; le second dans le gène HLA-DQA1 (locus 6p21 du) codant un antigène de classe II du complexe HLA associé à l’induction de la réponse immune (présentation de l’antigène). Dans cette étude, le risque relatif de développer une glomérulonéphrite extramembraneuse primaire en cas d’homozygotie pour les deux allèles à risque est de 78,5 (intervalle de confiance à 95 % : 34,6-178,2).
Plus récemment, une étude pangénomique réunissant des populations asiatique, européenne et nord-américaine, a identifié deux nouveaux locus à risque, dans les gènes NFKBA et IRF4, tous deux participant à la régulation immunitaire. Différents allèles à risque sur le locus HLA ont également été caractérisés, mais distincts selon l’ethnicité, suggérant une variabilité des facteurs déclenchants selon les populations.
Ces avancées récentes ouvrent ainsi des voies d’investigation des mécanismes moléculaires contribuant à la genèse de la glomérulonéphrite.
Diagnostic et évolution naturelle
La glomérulonéphrite extramembraneuse est habituellement révélée par un syndrome néphrotique, défini par une protéinurie supérieure à 3,5 g/24 h, une hypoalbuminémie inférieure à 30 g/L, cliniquement symptomatique sous forme d’un syndrome œdémateux périphérique d’importance variable.1 Ce tableau est parfois associé à une hématurie microscopique, une insuffisance rénale, voire une complication thrombotique inaugurale, en particulier une thrombose veineuse rénale alors responsable de lombalgies et d’hématurie macroscopique et parfois compliquée d’embolie pulmonaire.
Une révolution diagnostique a été apportée par les anticorps anti-PLA2R, recherchés idéalement à la fois par un test ELISA et un test d’immunofluorescence indirecte, ce dernier ayant la meilleure sensibilité. La
Une fois le diagnostic établi, l’enquête causale repose en partie sur la nature de l’antigène si celui-ci est identifié. Néanmoins, si certaines associations préférentielles entre antigènes cibles et causes sont retrouvées, il n’existe pas de lien univoque entre celles-ci (
Le bilan à la recherche d’une cause comporte la réalisation de sérologies infectieuses, la recherche de critères cliniques ou biologiques de maladie auto-immune, une anamnèse détaillée concernant d’éventuelles expositions médicamenteuses ou toxiques. Le dépistage d’une néoplasie solide ou hématologique est indispensable et doit, a minima, comprendre les examens de dépistage recommandés selon l’âge, le genre, les facteurs de risques et d’éventuels symptômes d’appel. Outre les cas rares de glomérulonéphrites extramembraneuses paranéoplasiques, la fréquence de détection d’une pathologie tumorale est environ 2 fois supérieur à celui de la population générale dans les années qui suivent le diagnostic de la glomérulonéphrite.
L’évolution spontanée est très variable : en l’absence de traitement spécifique, environ 20 % des patients ont à 5 ans une rémission complète, 40 % une rémission partielle, 40 % développent une insuffisance rénale chronique et 10 à 20 % évoluent vers l’insuffisance rénale terminale nécessitant un traitement de suppléance rénale.
Les facteurs prédictifs d’une évolution défavorable de la fonction rénale sont l’âge, le sexe masculin, l’altération du débit de filtration glomérulaire au moment du diagnostic, un taux élevé d’auto-anticorps circulants lors du diagnostic, des signes histologiques d’atteinte chronique du parenchyme rénal (fibrose interstitielle, atrophie tubulaire, lésions vasculaires), et une élévation de l’excrétion urinaire de protéines de bas poids moléculaire d’origine tubulaire. Au cours de l’évolution, l’absence de rémission du syndrome néphrotique ou la persistance d’un titre élevé d’auto-anticorps sont également déterminantes pour le devenir rénal.1
Traitement
Une fois le diagnostic de glomérulonéphrite extramembraneuse établi, le traitement standard associe un inhibiteur du système rénine-angiotensine à visée antiprotéinurique avec une cible tensionnelle inférieure à 130/80 mmHg, un traitement diurétique associé à un régime désodé (NaCl < 2 g/j) jusqu’à régression complète du syndrome œdémateux, éventuellement une statine en raison de la dyslipidémie secondaire au syndrome néphrotique, ainsi qu’une anticoagulation prophylactique à dose curative en présence d’un risque thrombotique majoré (hypoalbuminémie < 25 g/L). En cas de glomérulonéphrite secondaire, le traitement de la cause s’associe au traitement symptomatique.1
Le taux de rémissions spontanées étant important, l’indication d’un traitement immunosuppresseur ne s’envisage qu’en présence d’un syndrome néphrotique avec complications sévères (embolie pulmonaire, insuffisance rénale rapidement progressive), et/ou en l’absence de rémission au moins partielle voire complète 3 à 6 mois après l’initiation du traitement conservateur standard, ou en présence d’un titre d’anticorps anti-PLA2R très élevé.1 Le choix du protocole thérapeutique reste à la décision du néphrologue parmi les molécules reconnues efficaces, à savoir l’association cyclophosphamide/corticostéroïdes, les anti-calcineurines et le rituximab. Ce dernier, un anticorps monoclonal anti-CD20 dirigé contre les lymphocytes B, a pris une place grandissante ces dernières années, avec une efficacité globalement comparable à celles des autres molécules et un excellent profil de tolérance. En effet, comme rapporté par plusieurs essais thérapeutiques, notamment les essais GEMRITUX en France9 et MENTOR aux États-Unis,10 le rituximab permet d’obtenir 60 à 70 % de rémission, taux comparable à celui observé avec l’association cyclophosphamide-corticostéroides, standard de traitement historique, et une rémission plus prolongée que les anticalcineurines dont l’arrêt s’accompagne d’un taux élevé de rechutes.10 En outre, les risques carcinologiques et infectieux du rituximab en monothérapie sont moindres que l’association cyclophosphamide-corticostéroïdes, avec en outre l’absence de toxicité rénale contrairement aux anticalcineurines. Toutefois, les essais randomisés plus récents STARMEN et RI-Cyclo (sous presse) redonnent une place au cyclophosphamide. Une optimisation des thérapeutiques reste nécessaire afin d’atteindre un pourcentage de rémissions plus élevé et sans rechute, notamment grâce à une personnalisation des thérapeutiques existantes et/ou l’utilisation de nouveaux anticorps anti-lymphocyte B ou molécules ciblant les plasmocytes.
L’objectif thérapeutique est la préservation de la fonction rénale qui passe par l’obtention d’une rémission, au minimum partielle (protéinurie entre 0,3 et 3,5 g/j avec une réduction > 50 % par rapport à la valeur initiale) ou idéalement complète (protéinurie < 0,3 g/j). La rémission immunologique, définie par la disparition des anticorps anti-PLA2R circulants, est le marqueur le plus précoce de l’efficacité thérapeutique. L’évolution du débit de filtration glomérulaire et la présence de signes d’atteinte tubulaire sont aussi des facteurs importants dans le suivi du patient. La survenue de complications liées aux molécules immunosuppressives, notamment infectieuses ou néoplasiques, doit être considérée dans le suivi des patients atteints de glomérulonéphrite extramembraneuse.
Transplantation rénale
La greffe rénale est un moyen de suppléance rénale efficace pour les patients atteints de glomérulonéphrite extramembraneuse avec insuffisance rénale terminale. La présence d’auto-anticorps circulants anti-PLA2R lors de la transplantation est associée à la récidive de la glomérulonéphrite sur le greffon dans environ la moitié des cas dans l’année suivant la transplantation, réduisant la survie du greffon, et justifiant l’importance d’une rémission immunologique préalable à la transplantation.1
Il existe par ailleurs des cas de glomérulonéphrites extramembraneuses post-transplantation de novo sérologiquement négatives, compliquant environ 2 % des greffes rénales pour d’autres néphropathies. Leur nature reste mal comprise et leur évolution est fréquemment infraclinique.1
Conclusion
Malgré sa rareté, le diagnostic de glomérulonéphrite extramembraneuse ne doit pas être méconnu du médecin en première ligne en raison des complications sévères qui peuvent y être associées, à court et long terme, et de la nécessité d’une prise en charge néphrologique spécialisée.
Objet d’un champ de recherche en pleine ébullition, notamment grâce aux techniques modernes de protéomique, la liste des nouvelles molécules identifiées comme impliquées directement dans la physiopathologie de cette pathologie s’allonge à une vitesse impressionnante. Grâce également à plusieurs essais thérapeutiques majeurs et au monitorage des anticorps anti-PLA2R pour le suivi de l’efficacité des traitements, la dernière décennie a constitué une révolution dans la compréhension et la prise en charge de cette maladie, qui va se poursuivre dans les années à venir.
1. Couser WG. Primary Membranous Nephropathy. Clin J Am Soc Nephrol 2017;12:983-97.
2. Debiec H, Guigonis V, Mougenot B, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med 2002;346:2053-60.
3. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009;361:11-21.
4. Hanset N, Aydin S, Demoulin N, et al. Podocyte antigen staining to identify distinct phenotypes and outcomes in membranous nephropathy: a retrospective multicenter cohort study. Am J Kidney Dis 2020;76:624-35.
5. Sethi S, Debiec H, Madden B, et al. Semaphorin 3B-associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int 2020;98:1253-64.
6. Floege J, Barbour SJ, Cattran DC, et al. Management and treatment of glomerular diseases (part 1): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int 2019;95:268-80.
7. Caza T, Hassen S, Dvanajscak Z, et al. NELL1 is a target antigen in malignancy-associated membranous nephropathy. Kidney Int 2020;S0085-2538(20)30956-X.
8. Gupta S, Köttgen A, Hoxha E, et al. Genetics of membranous nephropathy. Nephrol Dial Transplant 2018;33:1493-1502.
9. Dahan K, Debiec H, Plaisier E, et al. Rituximab for severe membranous nephropathy: a 6-month trial with extended follow-up. J Am Soc Nephrol 2017;28:348-58.
10. Fervenza FC, Appel GB, Barbour SJ, et al.Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy. N Engl J Med 2019;381:36-46.