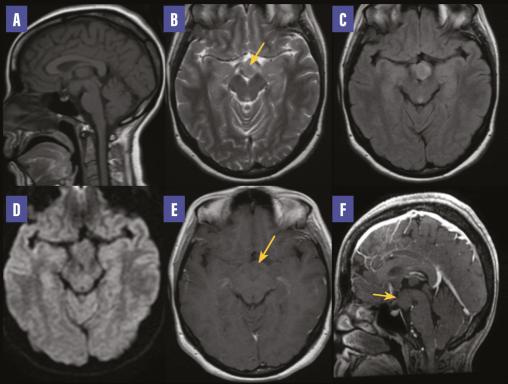
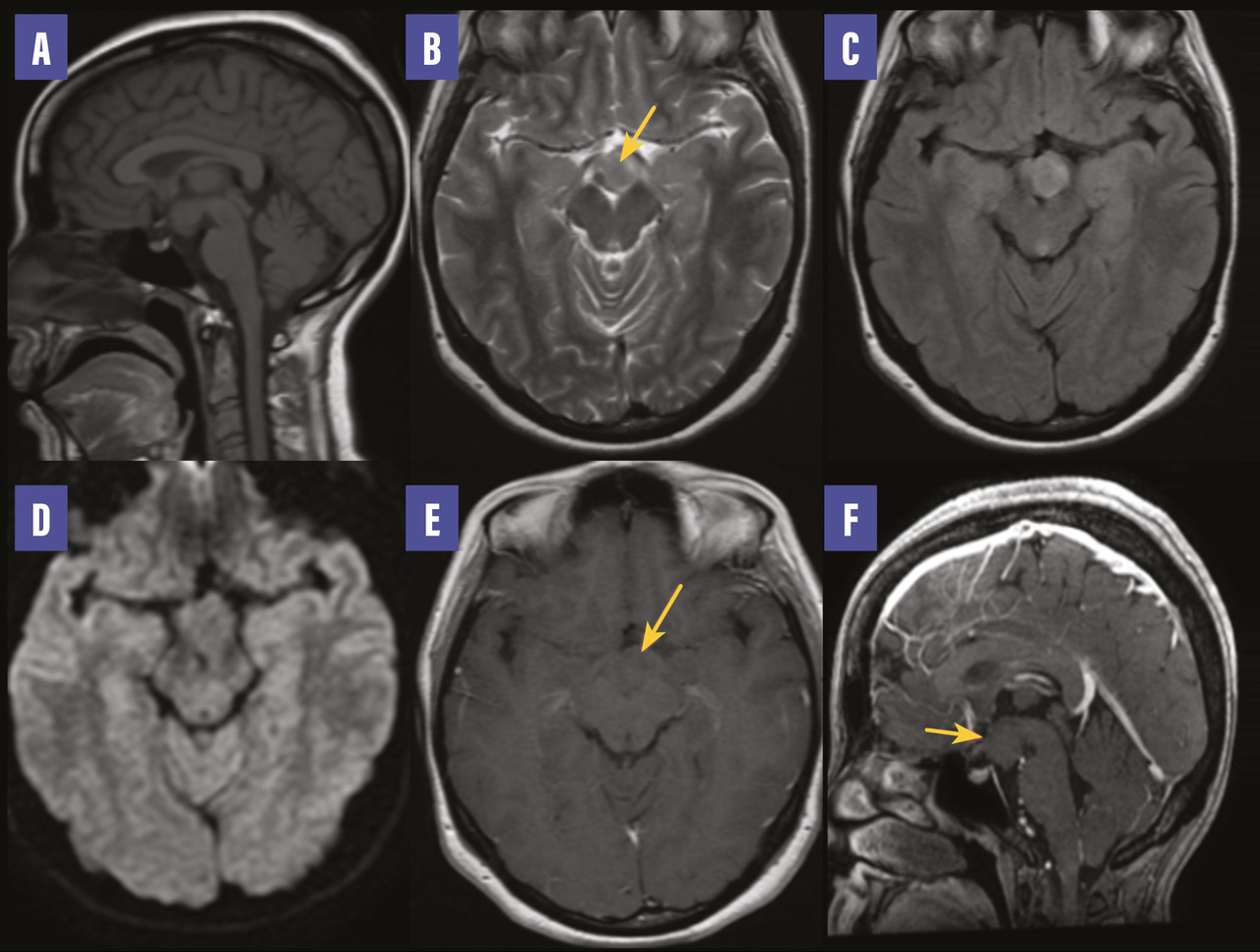
Une femme de 25 ans, mariée, deux enfants, sans antécédents particuliers, présente depuis un mois des crises épileptiques. À l’examen clinique, la patiente est consciente, stable sur le plan hémodynamique, normotendue, normocarde. Le bilan biologique est normal. Un électroencéphalogramme montre une activité épileptiforme bilatéralement synchrone et généralisée. La patiente, mise sous traitement antiépileptique, connaît une amélioration clinique. Par la suite, une imagerie par résonance magnétique cérébrale est réalisée dans le cadre du bilan étiologique ; elle montre une formation hypothalamique tissulaire en isosignal T1, hypersignal T2 et FLAIR, ne présentant pas de restriction sur la diffusion et ne se rehaussant pas après injection de gadolinium (figure ). Cette formation est centrée sur le tuber cinereum, avec respect de l’hypophyse. Devant cet aspect radiologique, le diagnostic d’hamartome hypothalamique est retenu.
L’hamartome hypothalamique est une malformation congénitale rare due à une prolifération anarchique de tissu glial et neuronal ectopique.1 Cette lésion bénigne non progressive ne dégénère pas.2 On en distingue deux types selon la localisation : le premier se situe au niveau hypothalamique près des corps mamillaires et est responsable de crises épileptiques gélastiques, le deuxième se trouve près de l’hypothalamus antérieur, du tuber cinereum et de la tige pituitaire et est habituellement associé à des signes de puberté précoce.2 Le traitement est médical en cas de puberté précoce et chirurgical en cas d’épilepsie réfractaire (résection ou déconnexion). La radiochirurgie par Gamma Knife peut également être proposée.1
Références
1. Arita K, Kurisu K, Kiura Y, Iida K, Otsubo H. Hypothalamic hamartoma. Neurol Med Chir (Tokyo) 2005;45(5):221‑31.
2. Harrison VS, Oatman O, Kerrigan JF. Hypothalamic hamartoma with epilepsy: Review of endocrine comorbidity. Epilepsia 2017;58(Suppl 2):50‑9.
2. Harrison VS, Oatman O, Kerrigan JF. Hypothalamic hamartoma with epilepsy: Review of endocrine comorbidity. Epilepsia 2017;58(Suppl 2):50‑9.
Une question, un commentaire ?