Rang A :
Connaître la définition clinique et biologique de l’hypoglycémie chez l’enfant.
Identifier une urgence : savoir reconnaître un coma hypoglycémique.
Connaître les médicaments du diabète pouvant provoquer une hypoglycémie chez le diabétique.
Connaître les principales étiologies d’hypoglycémie organique.
Mesures urgentes chez l’enfant.
Rang B :
Connaître les mécanismes de régulation de la glycémie.
Connaître les signes cliniques de l’hypoglycémie : différencier les signes neuroglucopéniques et adrénergiques.
Introduction
Reconnaître une hypoglycémie chez l’enfant est fondamental, du fait de :
- son urgence thérapeutique : tout retard au traitement peut mettre en jeu le pronostic (avant tout neurologique) ;
- son urgence étiologique : certaines causes exposent en plus du risque de récidive à un risque vital.
Ainsi les mesures urgentes devant une hypoglycémie sont de la corriger rapidement et efficacement (resucrage) et de réaliser un bilan sanguin au moment même de l’hypoglycémie (si l’état du patient le permet) afin de poser un diagnostic étiologique précoce.
Définition de l’hypoglycémie
L’hypoglycémie chez l’enfant est définie par une glycémie veineuse inférieure ou égale à 0,50 g/L (2,75 mmol/L). Ce seuil diffère dans trois cas particuliers :
- glycémie veineuse inférieure ou égale à 0,70 g/L (3,85 mmol/L) chez l’enfant diabétique traité par un médicament hypoglycémiant ;
- glycémie veineuse inférieure ou égale à 0,60 g/L (3,3 mmol/L) chez l’enfant porteur d’une maladie à risque d’hypoglycémie déjà diagnostiquée et autre que le diabète ;
- glycémie veineuse inférieure ou égale à 0,40 g/L (2,2 mmol/L) chez le nouveau-né durant les 48 premières heures de vie.
Physiopathologie
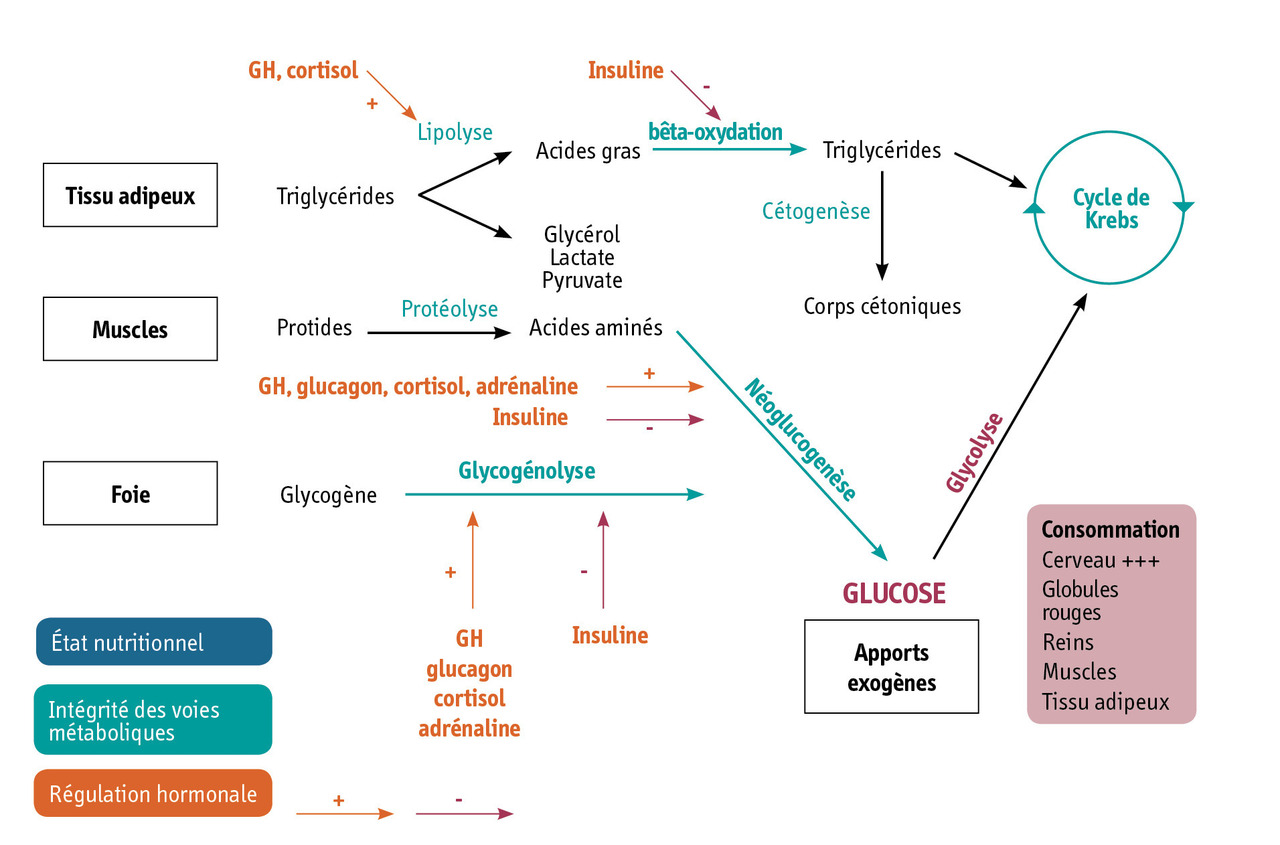
Acteurs de l’homéostasie glucidique
Le glucose est le substrat énergétique prédominant de l’enfant et de l’adulte. Chez l’enfant en bonne santé, l’homéostasie glucidique dépend :
- de l’intégrité du système hormonal, qui participe à la régulation des substrats mobilisés intervenant dans la synthèse du glucose. Il existe une seule hormone hypoglycémiante : l’insuline. Les autres hormones sont hyperglycémiantes : l’hormone de croissance, le cortisol, les catécholamines, le glucagon ;
- de l’intégrité des voies métaboliques (système enzymatique) intervenant dans la glycogénogenèse, la glycogénolyse et la néoglucogenèse, ainsi que des voies métaboliques permettant la disponibilité des autres substrats énergétiques impliqués dans l’oxydation et le stockage. Le foie est le principal siège de ces différentes voies métaboliques, et son intégrité est essentielle dans l’homéostasie glucidique ;
- de l’état nutritionnel et de la disponibilité suffisante en glycogène, en substrats de la néoglucogenèse (acides aminés, glycérol, lactate) et en lipides (acides gras libres) ;
- de la consommation de glucose par l’organisme (en particulier 3 fois plus importante chez le nouveau-né que chez l’adulte).
Tout dysfonctionnement au niveau de ces différentes interactions métaboliques peut être responsable d’une hypoglycémie. L’ensemble des facteurs contribuant à l’homéostasie glycémique de l’enfant est représenté dans la
Mise en jeu des différents acteurs de l’homéostasie glucidique : hypoglycémie du jeûne court ou du jeûne long ?
À l’état nourri, sous l’influence de l’insuline sécrétée par les cellules bêtapancréatiques en réponse à l’augmentation de la glycémie, le glucose ingéré est utilisé et stocké sous la forme de glycogène (glycogénogenèse) dans le foie et les muscles.
Au cours d’un jeûne court (3-12 heures), la glycémie est maintenue grâce à la mise en jeu de la dégradation du glycogène (glycogénolyse), stimulée par les hormones hyperglycémiantes.
Au cours d’un jeûne prolongé (> 12 heures), la glycémie est maintenue grâce à la néoglucogenèse alimentée par la protéolyse et la lipolyse. Les acides gras libres augmentés permettent également la production de corps cétoniques, substrats alternatifs au glucose.
La connaissance de la chronologie de la mise en jeu des différents acteurs de l’homéostasie glucidique et de la participation des différents substrats et hormones au cours du jeûne (
Absence d’acétonémie ou d’acétonurie : une information primordiale dans la démarche étiologique
Les corps cétoniques (acétoacétate, mesuré par la bandelette urinaire, et bêta-hydroxybutyrate, mesuré par la bandelette capillaire) sont des produits de la cétogenèse hépatique, à partir de la bêta-oxydation des acides gras libérés par la lipolyse. L’existence d’une acétonémie ou d’une acétonurie est le signe d’une bonne stimulation de la bêta-oxydation des acides gras et du bon fonctionnement de cette dernière. En situation d’hypoglycémie, l’absence d’acétonémie ou d’acétonurie peut donc avoir deux explications majeures :
– une inhibition forte de l’oxydation des acides gras par un hyperinsulinisme (endogène ou exogène) ;
– un défaut de synthèse des corps cétoniques par défaut enzymatique de l’oxydation des acides gras (déficit de la bêta-oxydation des acides gras).
Les corps cétoniques étant des substrats énergétiques alternatifs au glucose, en particulier pour le cerveau, les séquelles neurologiques sont plus sévères chez les nouveau-nés porteurs de ces deux pathologies (hypoglycémies hypocétoniques) que dans d’autres pathologies dans lesquelles cette voie n’est pas impactée (hypoglycémies hypercétoniques).
Reconnaître l’hypoglycémie : diagnostic positif
Chez l’enfant comme chez l’adulte, l’hypoglycémie est suspectée devant deux types de signes cliniques.
Les symptômes de la réaction adrénergique (neurovégétative) liés à l’activation neuronale sympathique, non spécifiques de l’hypoglycémie, et qui peuvent être absents lorsque les épisodes d’hypoglycémie se répètent ou en cas de neuropathie végétative : pâleur, sueurs, tachycardie, palpitations, tremblements, irritabilité, sensation de faim et asthénie.
Les symptômes de la réaction neuroglucopénique traduisant la privation cérébrale en glucose : céphalées, troubles de la concentration, troubles visuels, troubles d’élocution, hallucinations, confusion, troubles du comportement, vertiges, lipothymie ou malaise, déficit neurologique, convulsions, signes pyramidaux, dysesthésie, somnolence, hypothermie et coma. Tout trouble de la conscience ou tout syndrome neurologique aigu doit faire rechercher une hypoglycémie, jusqu’à preuve du contraire.
Chez le nourrisson, les signes cliniques sont fréquemment aspécifiques, pouvant faire évoquer d’autres pathologies (sepsis, pathologies cardiorespiratoires) : cyanose, détresse respiratoire, difficultés d’alimentation, irritabilité, léthargie, hypothermie. Tout symptôme de ce type chez le nouveau-né doit faire réaliser une glycémie capillaire.
La triade de Whipple (signes neuroglucopéniques ; glycémie veineuse < 0,50 g/L ; correction des symptômes après normalisation de la glycémie) permet le diagnostic d’hypoglycémie.
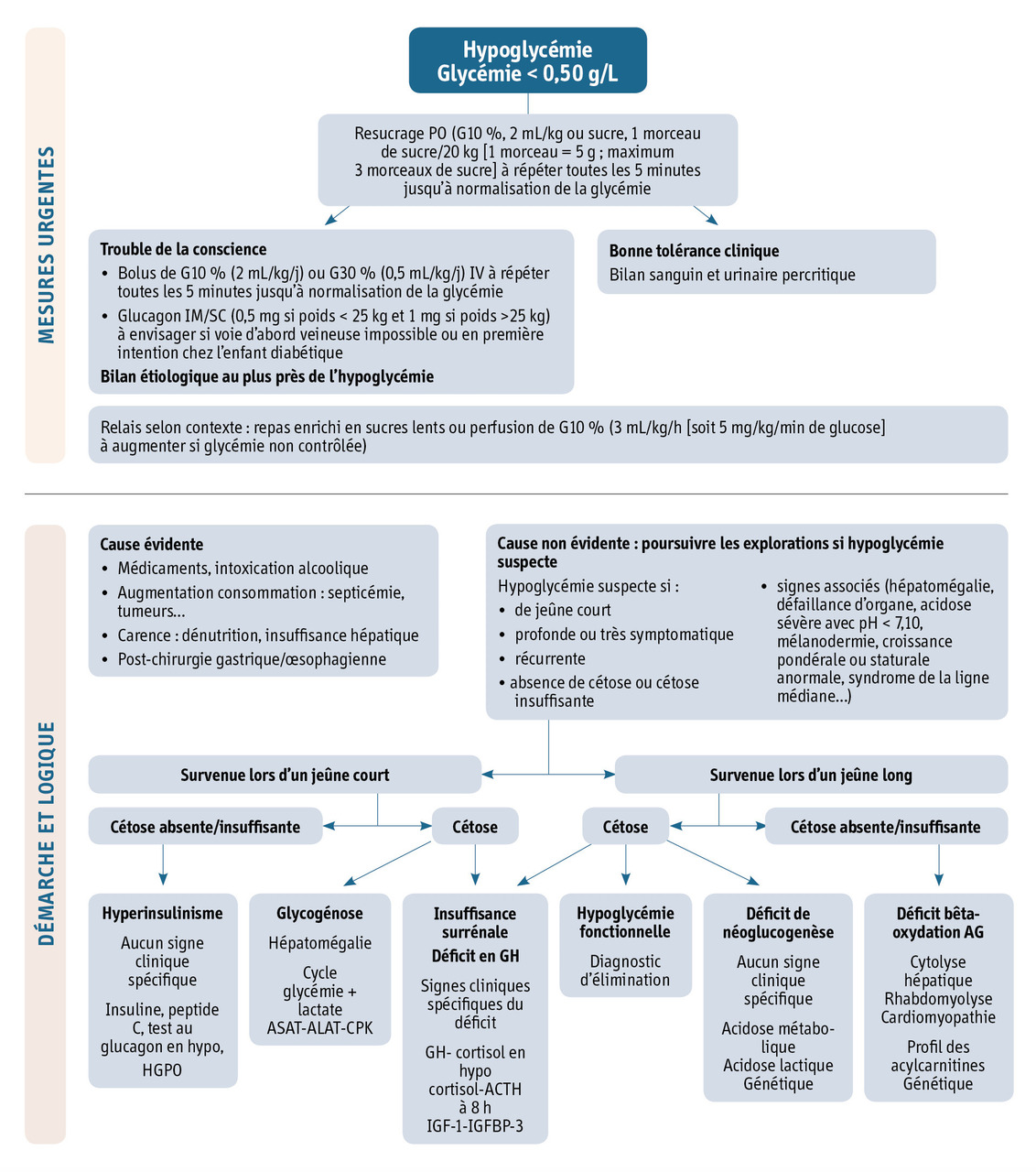
Prise en charge d’une hypoglycémie en urgence
Une glycémie capillaire doit être pratiquée en urgence devant tout signe évocateur d’hypoglycémie. La correction d’une hypoglycémie constitue une urgence thérapeutique. L’enquête étiologique (bilan sanguin et urinaire) doit être faite au plus proche de l’hypoglycémie, idéalement avant resucrage, sauf en cas d'urgence vitale ou d’enfant inconscient. La prise en charge initiale de l’hypoglycémie dépend donc de l’état du patient (
Enfant inconscient
L’enfant doit recevoir immédiatement un traitement visant à normaliser sa glycémie :
- bolus de solution glucosé en intraveineux (à répéter toutes les 5 minutes jusqu’à normalisation de la glycémie) : 2 mL/kg de solution glucosée 10 % (nouveau-né et nourrisson) ou 0,5 mL/kg de solution glucosée 30 % (enfant et adolescent) ;
- relais par solution glucosée 10 % en perfusion : 3 mL/kg/h (soit 5 mg/kg/min de glucose) à augmenter si glycémie non contrôlée ;
- glucagon en injection intramusculaire ou sous-cutanée à envisager si voie d’abord veineuse impossible (0,5 mg chez l’enfant de moins de 25 kg et 1 mg chez l’enfant de plus de 25 kg). Le glucagon permet la libération du glucose hépatique. Il est donc inefficace en cas de glycogénose ou d’hypoglycémie au jeûne prolongé. À l’inverse, son action est considérable si la cause est liée à une hyperinsulinémie. Chez le diabétique connu, il s’agit de la première mesure à réaliser au domicile par la famille.
Le bilan étiologique doit être réalisé après resucrage, mais au plus près de l’hypoglycémie (l’insuline et le peptide C ne seront pas interprétables dans cette situation).
Enfant conscient
Le bilan étiologique doit être réalisé avant resucrage (prélèvement sanguin et urinaire percritique).
L’enfant doit recevoir rapidement un traitement visant à normaliser sa glycémie :
- nouveau-né et nourrisson : glucose 10 %, 2 mL/kg, par voie orale ;
- enfant et adolescent : sucre (1 morceau = 5 g) ; 1 morceau de sucre/20 kg (maximum 3 morceaux de sucre).
Après normalisation de la glycémie, il faut assurer en relais des apports prolongés en glucides pour éviter une récidive immédiate d’hypoglycémie par l’intermédiaire soit d’un repas enrichi en sucres lents, soit d’une perfusion de solution glucosée à 10 % (3 mL/kg/h [soit 5 mg/kg/min de glucose]) selon l’état du patient.
Démarche diagnostique devant une hypoglycémie : enquête étiologique
Une hypoglycémie peut être expliquée :
- par la défaillance d’un acteur de l’homéostasie glucidique : dysfonctionnement hormonal (hyperinsulinisme, déficit en hormone de croissance, déficit en cortisol), défaut des voies métaboliques (glycogénose, défaut de la néoglucogenèse, trouble de la bêta-oxydation, hépatopathie) ;
- par un défaut d’apports glucidiques (dénutrition, anorexie…) ou une anomalie de l’absorption digestive ;
- par une augmentation importante des besoins (sepsis, état de choc, brûlures, tumeurs) ;
- par une intoxication médicamenteuse ou la prise de toxiques (alcool).
L’hypoglycémie fonctionnelle avec cétose est une situation fréquemment observée mais qui doit rester un diagnostic d’élimination. Les causes d’hypoglycémie sont donc extrêmement variées et diffèrent selon l’âge. Leurs caractéristiques principales sont rassemblées dans le
Des données simples anamnestiques (jeûne court/long, contexte), cliniques (hépatomégalie) et biologiques (glycémie, lactacidémie, cétonurie, hormonologie, acylcarnitine) conduisent à un diagnostic dans la majorité des cas (
La démarche étiologique commence par l’élimination d’une cause d’hypoglycémie évidente car survenant dans un contexte particulier :
- augmentation de la consommation de glucose : septicémie ; pathologie aiguës, tumeurs ;
- carence en glycogène ou en substrat pour la néoglucogenèse : dénutrition sévère, insuffisance hépatique profonde aiguë ou chronique ;
- patients avec antécédent de chirurgie gastrique ou œsophagienne (dumping syndrome) ;
- fuite urinaire de glucose : tubulopathie ;
- intoxication alcoolique ou prise de médicaments (insuline, sulfamides ;
- bêtabloquants, certaines chimiothérapies, sevrage brutal d’une corticothérapie prolongée et/ou à forte dose).
La suite de la démarche étiologique comprend le recueil des circonstances de survenue (âge de l’enfant ; horaire de survenue : jeûne court ou long ; épisode unique ou récidive) et la réalisation d’un examen clinique complet, avec une attention particulière sur l’examen neurologique, la recherche d’une hépatomégalie, orientant vers une glycogénose, la recherche d’un retard de croissance, d'un micropénis, d'un ictère prolongé ou d'une anomalie de la ligne médiane (fente labiopalatine, par exemple), orientant vers un hypopituitarisme avec déficit en hormone de croissance et la recherche d’une mélanodermie, orientant vers une insuffisance surrénalienne primaire.
La cause peut être également diagnostiquée à la suite du prélèvement percritique. Ce bilan comporte :
- au moment de l’hypoglycémie (avant resucrage) :
- glycémie veineuse ;
- GH, cortisol, insuline et peptide C ;
- et en cas d’hypoglycémie au jeûne court : test au glucagon en hypoglycémie, 1 mg en sous-cutané, et mesure de glycémie capillaire et veineuse à 5, 10, 15 et 30 minutes ; le test est positif lorsque l’augmentation de glycémie est supérieure à 0,30 g/L et montre donc une réponse forte ;
- au plus proche de l’hypoglycémie (éventuellement juste après resucrage) :
dans le sang :
- gaz du sang (pH, réserve alcaline) ;
- lactates ;
- carnitine libre et totale ; profil des acylcarnitines plasmatiques ;
- bêta-hydroxybutyrate (= corps cétoniques) ;
- acides gras libres ;
- ionogramme sanguin ; uricémie, triglycéridémie, transaminases, CPK ;
- ammoniémie ;
- IGF-1 ;
- dosage de toxiques et de médicaments :
dans les urines :
- bandelette urinaire : recherche de cétonurie et glycosurie ;
- chromatographie des acides organiques urinaires ;
- recherche de toxiques.
À ce stade, en l’absence de diagnostic au terme des premiers résultats, ou si les prélèvements en hypoglycémie n’ont pas pu être réalisés, des explorations complémentaires peuvent être conduites dans des services spécialisés (service d’endocrinologie ou de maladies métaboliques pédiatriques), en particulier quand les hypoglycémies sont suspectes. Les hypoglycémies peuvent être considérées comme suspectes quand elles sont inattendues (hypoglycémies pour un jeûne pas assez long pour l’âge : < 6 heures à la naissance, < 12 heures entre 1 et 5 ans et < 24 heures entre 5 et 10 ans ; après 10 ans, il ne devrait plus y avoir d’hypoglycémie de jeûne), inappropriées (hypoglycémies très symptomatiques ou très profondes; insuffisance ou absence de cétose), ou accompagnées de signes associés (hépatomégalie, défaillance d’organe, acidose sévère avec pH < 7,10, mélanodermie, croissance pondérale ou staturale anormale…).
En cas d’hypoglycémies suspectes, les explorations complémentaires doivent être réalisées en fonction de l’horaire de survenue du jeûne.
Les hypoglycémies du jeûne court sont par définition anormales, et nécessitent des explorations complémentaires : un cycle de glycémie-lactatémie avant et après un repas (prélevé sur une voie veineuse périphérique juste avant le repas, et 1 heure après le prélèvement préprandial) pendant 24 heures, et/ou une hyperglycémie provoquée par voie orale (HGPO) avec mesure de la glycémie et de l’insuline pendant 2 à 4 heures. En cas d’hypoglycémie décelée : un bilan sanguin et urinaire percritique et un test au glucagon devront être réalisés. Les diagnostics à évoquer sont l’hyperinsulinisme (hypoglycémies sans horaire, quelle que soit la durée du jeûne, y compris en post-prandial, absence de cétose, nécessité d’apports supraphysiologiques en glucose), une glycogénose (présence d’une hépatomégalie), un panhypopituitarisme avec déficit en GH et éventuellement en cortisol (anomalie de la ligne médiane, retard de croissance, micropénis, ectopie testiculaire, ictère prolongé, déficit thyréotrope associé avec T4 basse). Au cours d’une gastroentérite, entre la diminution de l’alimentation, la malabsorption, et les vomissements, toute hypoglycémie doit être considérée comme étant de jeûne long et confirmée biologiquement par la présence dans ce contexte d’une forte cétose. L’enfant sera alors perfusé jusqu’à la disparition complète de la cétose et un retour à une alimentation normale. Après arrêt de la perfusion, il est conseillé de réaliser un cycle glycémique sur 24 heures sous une alimentation normale pour vérifier l’absence de récidive d’hypoglycémie du jeûne court.
En cas d’hypoglycémie du jeûne long, un déficit hormonal, un défaut de la néoglucogenèse, un déficit de la bêta-oxydation des acides gras doivent être évoqués. Une cétose doit toujours être recherchée : l’absence de cétose du jeûne long est anormale et oriente vers un déficit de la bêta-oxydation des acides gras. À l’inverse, les déficits de la néoglucogenèse s’accompagnent d’une forte cétose et d’une acidose sévère. La présence d’une hyperlactatémie au moment de l’hypoglycémie signe un dysfonctionnement de la néoglucogenèse. En l’absence de diagnostic, il ne faut réaliser une épreuve de jeûne en milieu spécialisé (durée adaptée à l’âge de l’enfant, au maximum 24 heures) qu’après avoir exclu un déficit de la bêta-oxydation des acides gras et une insuffisance surrénalienne par les dosages correspondants (soit percritiques au moment de l’hypoglycémie, soit le matin à jeun). Au cours de l’épreuve de jeûne, des dosages hormonaux (insuline, peptide C, cortisol, GH) et métaboliques (profil des acylcarnitines plasmatiques, lactatémie, 3-bêta-hydroxybutyrate, chromatographie des acides organiques urinaires) sont prélevés régulièrement pendant l’épreuve et au cours de l’éventuelle hypoglycémie, afin de vérifier l’adaptation des sécrétions hormonales au jeûne prolongé et l’intégrité de la néoglucogenèse. Le diagnostic d’hypoglycémie fonctionnelle ou hypoglycémie hypercétonique idiopathique est un diagnostic d’élimination. Ce diagnostic, fréquent, bénin et survenant pour des jeûnes très prolongés chez des enfants de moins de 10 ans, associe une hypoglycémie peu profonde (> 0,40 g/L), peu symptomatique, survenant au jeûne très prolongé, une forte cétose et l’absence d’élément orientant vers une maladie hypoglycémiante sous-jacente.
Points clés à propos de certaines causes
Hypoglycémie chez le patient diabétique
Chez les patients diabétiques (diabète de type 1 dans la majorité des cas chez l’enfant), la survenue d’une hypoglycémie est très généralement d’origine iatrogène et favorisée par la prise d’insuline (médicament le plus utilisé chez l’enfant diabétique) ou de sulfamides hypoglycémiants (utilisés dans certains cas de diabètes rares monogéniques). La prévention des hypoglycémies est essentielle chez l’enfant diabétique et repose sur une éducation du patient et de son entourage à la surveillance des glycémies (mesures capillaires ou interstitielles continues), la bonne technique d’injection (prévention des lipodystrophies), l’ajustement de l’insulinothérapie (en fonction de la glycémie, de la quantité de glucides du repas, d’une activité physique prévue), la reconnaissance des signes d’hypoglycémie et la conduite à tenir en cas de survenue. En l’absence de trouble de la conscience, l’enfant doit se resucrer par voie orale (5 g de sucre/20 kg, maximum 3 morceaux de sucre). En présence de trouble de la conscience ou en cas d’incapacité de prise de sucre par voie orale, l’injection de glucagon par voie intramusculaire ou sous-cutanée (et bientôt nasale) est la première mesure à réaliser au domicile par la famille. La répétition d’hypoglycémies ou la survenue d’hypoglycémies particulièrement sévères doit faire rechercher une maladie associée, en particulier une insuffisance surrénalienne auto-immune ou une malabsorption par maladie cœliaque, plus fréquentes dans ce contexte. Enfin, il faut être vigilant à l’administration volontaire d’insuline en excès, tout particulièrement chez le grand enfant et l’adolescent.
Hyperinsulinisme congénital
L’hyperinsulinisme congénital est un dysfonctionnement primaire de la cellule bêta du pancréas conduisant à une sécrétion inappropriée d’insuline, quelle que soit la glycémie. Il est d’origine génétique et peut soit impliquer uniquement la cellule bêta du pancréas (hyperinsulinisme congénital isolé) soit s’inscrire dans le cadre d’un syndrome (hyperinsulinisme syndromique). Les hypoglycémies peuvent se révéler à tous les âges.
Chez le nouveau-né et le nourrisson, l’hyperinsulinisme congénital dans sa forme la plus sévère peut être responsable d’hypoglycémies sévères récidivantes dès les premières heures de vie, avec convulsions ou coma. L’élément orientant vers un hyperinsulinisme congénital est la nécessité de besoins d’apports très élevés de glucose pour éviter la récidive d’hypoglycémie. Cette pathologie est à distinguer de l’hyperinsulinisme transitoire à la naissance, lié à un diabète maternel mal équilibré (nouveau-né macrosome) ou secondaire à une asphyxie périnatale.
Chez l’enfant plus grand, l’hyperinsulinisme doit être évoqué devant des hypoglycémies récurrentes sans horaire fixe, sans cétose, avec insulinémie inadaptée au moment de l’hypoglycémie, et ponctuellement corrigées par le test au glucagon. Il est à distinguer de l’insulinome, qui est une tumeur neuroendocrine pancréatique exceptionnelle chez l’enfant. Une cause exogène, en particulier l’administration occulte d’insuline ou de sulfamides hypoglycémiants pratiquée par un proche, ne doit pas être méconnue et doit faire évoquer une maltraitance et en particulier un syndrome de Münchhausen par procuration.
Étant donné que l’hyperinsulinisme inhibe la production de corps cétoniques (substrats énergétiques alternatifs au glucose, en particulier pour le cerveau), il existe en particulier chez le nouveau-né un risque de dommages cérébraux irréversibles : son diagnostic et son traitement sont donc des extrêmes urgences.
POINTS FORTS À RETENIR
Tout trouble de la conscience ou tout syndrome neurologique aigu doit faire rechercher une hypoglycémie, jusqu’à preuve du contraire.
La correction d’une hypoglycémie constitue une urgence thérapeutique.
La triade de Whipple (signes neuroglucopéniques; glycémie veineuse < 0,50 g/L; correction des symptômes après normalisation de la glycémie) permet d’évoquer le diagnostic d’hypoglycémie.
L’enquête étiologique est une urgence car certaines causes exposent, en plus du risque de récidive, à un risque vital.
L’enquête étiologique débute dès le diagnostic par la réalisation d’un bilan percritique avant le resucrage si l’état de l’enfant le permet.
La démarche étiologique repose sur l’analyse de données simples anamnestiques (jeûne court/long, contexte), cliniques (hépatomégalie) et biologiques (glycémie, lactacidémie, cétonurie, hormonologie, acylcarnitine).
Une cause exogène, en particulier l’administration occulte d’insuline ou de sulfamides hypoglycémiants pratiquée par un proche, ne doit pas être méconnue et doit faire évoquer un syndrome de Münchhausen par procuration.
Chez le diabétique, l’hypoglycémie définie par une glycémie capillaire < 70 mg/dL est iatrogène (surdosage en insuline, voire en sulfamides), et sa prévention repose sur l’éducation thérapeutique du patient et de son entourage.
Hypoglycémie chez l’adulte et l’enfant
Deux grandes situations d’hypoglycémie pourraient faire l’objet d’un dossier progressif :
Une hypoglycémie chez un diabétique de type 1 connu : le cas clinique pourrait débuter par une découverte de diabète suivie par la survenue plusieurs semaines plus tard d’hypoglycémies. L’étudiant doit connaître les gestes d’urgence en cas d’hypoglycémie (resucrage, glucagon). En plus des causes d’hypoglycémies les plus fréquentes chez le diabétique (lipodystrophies, surdosage, mauvais comptage des glucides, absence d’ajustement de l’insulinothérapie selon la glycémie ou le sport), l’étudiant devra évoquer une maladie associée fréquente du diabète de type 1 : l’insuffisance surrénalienne auto-immune ou la maladie cœliaque. L’étudiant devra connaître la nécessité d’une information et d’une éducation du patient et de son entourage vis-à-vis de ce risque.
Une hypoglycémie chez un enfant sans antécédent, suspectée devant un malaise, une perte de connaissance ou tout signe neurologique. L’étudiant devra évoquer une hypoglycémie, connaître les gestes d’urgence et débuter l’enquête étiologique. L’étudiant ne devra pas méconnaître une hypoglycémie médicamenteuse (pouvant faire évoquer un syndrome de Münchhausen par procuration), une insuffisance surrénalienne ou hypophysaire.
Collège national des pédiatres universitaires et CNHUCP. 8e édition. 2021. Item 240 : Hypoglycémie chez l’adulte et chez l’enfant
Collège des enseignants d’endocrinologie, diabète et maladies métaboliques. 4e édition, 2018. Item 238 : Hypoglycémie chez l’adulte et chez l’enfant.
Arnoux JB. Hypoglycémies de l’enfant (hors nouveau-né et diabètes). Pas à pas en pédiatrie.2019.
Thornton PS, Stanley CA, De Leon DD, Harris D, Haymond MW, Hussain K, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children; Pediatric Endocrine Society. J Pediatr 2015;167(2):238-45.
 Encadrés
Encadrés