L’électrophorèse des protides sériques (EPS) est un examen simple et peu coûteux. Elle permet l’analyse et la séparation des protéines sériques par migration dans un champ électrique en fonction de leur poids et de leur charge.
Quand demander une électrophorèse des protides sériques ?
Les indications à la réalisation d’une électrophorèse des protides sériques sont bien établies. La Haute Autorité de santé (HAS) a en effet publié des recommandations en 2017 : les signes d’appel cliniques, biologiques ou radiologiques y sont listés (
Extrêmement rare mais véritable urgence thérapeutique, le syndrome d’hyperviscosité fait partie des indications. Survenant en contexte d’hémopathie sous-jacente, il doit être suspecté en cas de saignements inexpliqués ou de symptômes neurologiques, respiratoires, cardiovasculaires. Son diagnostic repose sur un faisceau d’arguments et un avis d’experts. L’examen le plus spécifique en est le fond d’œil, qui peut mettre en évidence une dilatation des vaisseaux rétiniens, des hémorragies rétiniennes, des nodules cotonneux, des micro-anévrismes, voire une thrombose de la veine centrale de la rétine et un œdème papillaire.
Anomalies des protides autres que les gammaglobulines
L’hypoalbuminémie peut correspondre à une hémodilution, à une dénutrition profonde ou à une fuite urinaire importante (syndrome néphrotique).
L’hyper-alpha-2-globulinémie est un marqueur de syndrome inflammatoire.
Le bloc bêta-gamma, qui déforme la zone des bêta- et gammaglobulines, est un signe de cirrhose.
Quoique plus fréquent dans les gammaglobulines, un pic monoclonal peut migrer et apparaître dans les bêtaglobulines.
Trois types d’anomalies des gammaglobulines
Hypogammaglobulinémie
Elle se définit par une baisse des gammaglobulines contrôlée, inférieure à 5 g/L.2,3
Hypogammaglobulinémie primitive
Elle se révèle à la naissance ou plus tardivement. Les déficits néonataux (agammaglobulinémie du type maladie de Bruton, par exemple) en sont une des causes.
Plus fréquent, le déficit immunitaire commun variable (DICV) doit être évoqué devant des infections ORL et pulmonaires récidivantes, couplées à un défaut de réponse vaccinale. Son diagnostic est le plus souvent posé entre 20 et 30 ans, parfois chez le sujet bien plus âgé. Des infections digestives, des pathologies auto-immunes (thyroïdite, cytopénies) ou une granulomatose peuvent y être associées. Le traitement substitutif par immunoglobulines intraveineuses ou sous-cutanées améliore le pronostic.
Hypogammaglobulinémie secondaire
Elle peut être liée à différentes causes. De diagnostic exceptionnel ou évident, les carences nutritionnelles majeures (kwashiorkor), les entéropathies exsudatives, le syndrome néphrotique peuvent en être responsables.
Iatrogénique, elle est le plus souvent due aux antiépileptiques (carbamazépine, phénytoïne, clonazépam), aux cytotoxiques, aux biothérapies (rituximab), aux antipsychotiques (clozapine).
Les hémopathies lymphoprolifératives doivent être également évoquées systématiquement devant une hypogammaglobulinémie de l’adulte : leucémie lymphoïde chronique, lymphome non hodgkinien, myélome à chaîne légère. Représentant environ 10 % des myélomes, ce dernier peut se révéler initialement par une hypogammaglobulinémie isolée. Par définition, il ne se traduit pas par un pic monoclonal à l’électrophorèse des protides sériques. En revanche, la chaîne légère monoclonale (kappa ou lambda) peut être mise en évidence par la réalisation d’une électrophorèse urinaire (la recherche de protéinurie de Bence-Jones est obsolète) et surtout, à l’heure actuelle, par le dosage des chaînes légères sériques (augmentation significative d’une chaîne légère par rapport à l’autre).
Hypergammaglobulinémie polyclonale
Elle reflète une activation aspécifique des lymphocytes B, déformant « en dôme » la zone des gammaglobulines sur l’EPS. Le seuil à partir duquel les explorations sont nécessaires n’est pas consensuel, mais on considère comme anormal un taux supérieur à 25 ou 30 g/L.4,5
Les causes en sont nombreuses : hépatopathies chroniques (cirrhose alcoolique ou toxique, hépatites virales chroniques, hépatites auto-immunes, cholangite biliaire primitive, cholangite sclérosante, etc.), connectivites (lupus systémique, syndrome de Sharp, syndrome de Gougerot-Sjögren), infections chroniques (virus de l’immunodéficience humaine [VIH], endocardite, abcès profond, etc.), certaines parasitoses (leishmaniose, trypanosomiase, paludisme), pathologies hématologiques (lymphomes B et T, myélodysplasies comme la leucémie myélomonocytaire chronique).
La sarcoïdose peut également être révélée par une hypergammaglobulinémie polyclonale, ce qui justifie la réalisation systématique d’une imagerie thoracique, à la recherche d’adénopathies médiastinales.
Rarement, l’hypergammaglobulinémie polyclonale est révélatrice de tumeurs solides (carcinome hépatocellulaire, carcinome ovarien, carcinomes pulmonaires épidermoïdes, etc.).
D’autres causes sont possibles : maladies inflammatoires chroniques intestinales, thyroïdites auto-immunes ou encore le rare syndrome d’hyper-IgG4.
Enfin, il est classique de retrouver une hypergammaglobulinémie chez les sujets originaires d’Afrique subsaharienne, et ce indépendamment de parasitose sous-jacente.
Dysglobulinémie monoclonale
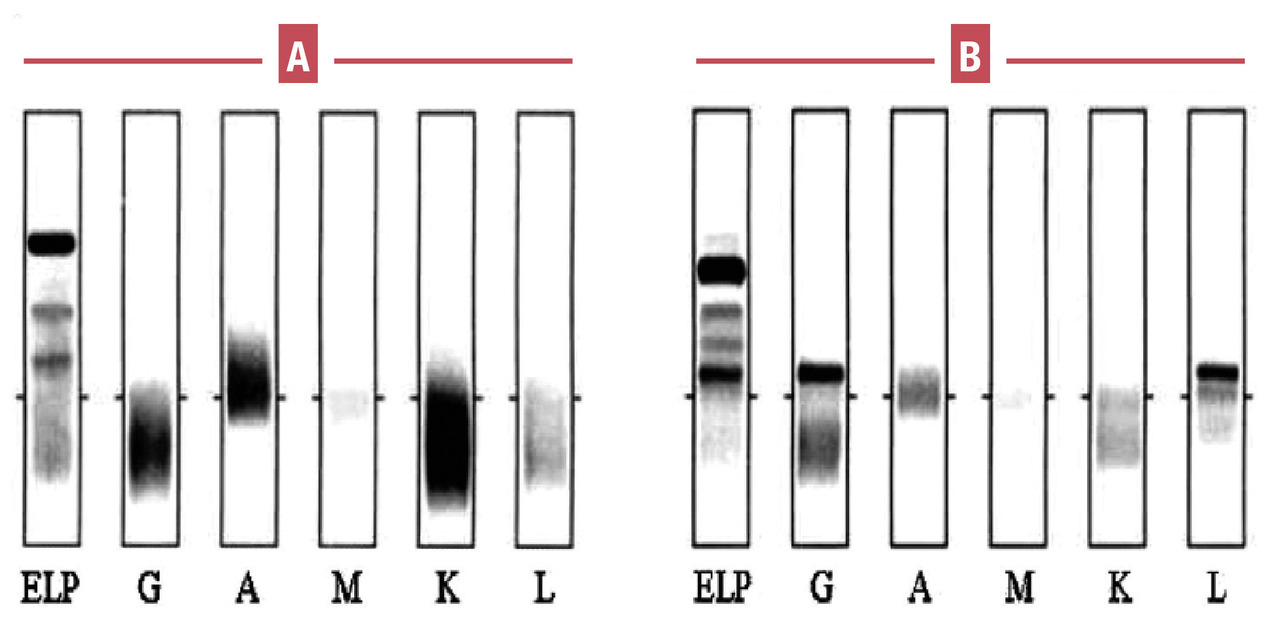
Elle correspond à une immunoglobuline constituée d’un seul type de chaîne lourde (alpha, gamma…) et d’un seul type de chaîne légère (kappa ou lambda). Elle témoigne de la prolifération d’un même clone cellulaire et se manifeste par un pic pointu, à base étroite, symétrique et homogène, migrant le plus souvent dans les gammaglobulines (IgG) [
Dysglobulinémie monoclonale satellite d’une autre pathologie
Toute situation infectieuse aiguë peut s’accompagner transitoirement d’une dysglobulinémie monoclonale qui disparaît avec l’infection. Il convient donc de contrôler l’EPS à distance.
Pour les infections chroniques par le VIH ou le virus de l’hépatite C, on l’observe dans 5 à 10 % des cas.
Parmi les connectivites, le syndrome de Gougerot-Sjögren s’assortit parfois d’une dysglobulinémie monoclonale, mais la possibilité d’un lymphome associé doit rendre prudent.
En dermatologie, elle peut s’observer dans les mucinoses et les dermatoses neutrophiliques.
Enfin, elle peut être présente dans toute situation post-greffe médullaire ou d’organes solides.
Dysglobulinémie monoclonale de signification indéterminée
On parle aussi de MGUS, pour monoclonal gammopathy of undetermined significance.6,7
Répondant à des critères diagnostiques stricts, les MGUS sont le plus souvent à IgG (70-75 %) ou IgM (15-20 %), et leur fréquence augmente avec l’âge, atteignant 7,5 % après 85 ans (
Les MGUS sont considérées comme des situations précancéreuses : le risque d’évolution maligne est de 1 % par an pour une MGUS IgG et de 3 à 5 % pour une MGUS IgM.
Les MGUS non IgM évoluent vers le myélome multiple, et les MGUS IgM vers la maladie de Waldenström. La surveillance à six mois, puis annuelle est donc impérative. Elle consiste en un examen clinique et un bilan biologique (EPS, calcémie, créatininémie, hémogramme). En cas d’apparition de signes cliniques, de progression de plus de 25 % du pic ou d’autres anomalies des examens biologiques, un avis spécialisé est nécessaire.
Dysglobulinémie monoclonale pouvant révéler un myélome
Il peut alors s’agir d’un pic IgG, IgA, IgD, d’une prolifération de chaîne légère ou, très exceptionnellement, d’un pic IgM.8,9 Les critères diagnostiques de la maladie ont été modifiés ces dernières années par l’apport de l’imagerie moderne, le dosage des chaînes légères et l’apparition du concept de « myélome indolent ».
Le myélome est ainsi actuellement défini par l’association de deux critères :
– une plasmocytose médullaire au myélogramme > 10 % ;
– et au moins un signe CRAB (
Le myélome indolent est, quant à lui, défini par un pic monoclonal supérieur à 30 g/L, une plasmocytose médullaire entre 10 et 60 %, l’absence de signes CRAB et un rapport des chaînes légères sériques équilibré.
Dysglobulinémie monoclonale correspondant à une pathologie lymphoproliférative
La dysglobulinémie monoclonale à IgM correspond le plus souvent à une pathologie lymphoproliférative. Ce peut être une maladie de Waldenström qui associe à l’IgM monoclonale une infiltration lymphoplasmocytaire avec adénopathies périphériques et hépatosplénomégalie. Le myélogramme montre une infiltration supérieure à 10 %. L’IgM étant une molécule pentavalente, le syndrome d’hyperviscosité (céphalées, troubles de conscience, hémorragies au fond d’œil) peut être révélateur. D’autres proliférations lymphocytaires à IgM sont possibles : leucémie lymphoïde chronique (avec hyperlymphocytose sanguine dont la clonalité est affirmée par l’immunophénotypage, éventuellement associée à des adénopathies et une splénomégalie), lymphomes non hodgkiniens, rarement myélodysplasies (leucémie myélomonocytaire chronique, par exemple).
Dysglobulinémies monoclonales pouvant avoir une activité pathogène propre
Dans ces cas, divers mécanismes sont en cause (pics souvent faibles) :
– l’immunoglobuline monoclonale peut s’attaquer préférentiellement à un organe, une cellule ou une protéine. Le rein est l’une des cibles (on parle de MGRS, pour monoclonal gammopathy of renal significance), avec des néphropathies tubulaires ou glomérulaires. Le nerf peut également être concerné : neuropathie avec des IgM (plus rarement IgG) actives contre des éléments du nerf périphérique (le plus souvent la gaine de myéline avec une activité anti-MAG [myelin-associated glycoprotein]). Le globule rouge peut être atteint : maladie des agglutinines froides, où le pic est actif contre une protéine de la membrane des globules rouges. Enfin l’immunoglobuline monoclonale peut toucher des composants lipidiques (cas des xanthomes normolipémiques) ;
– l’immunoglobuline monoclonale peut être l’un des éléments d’un syndrome. C’est le cas du syndrome de POEMS (polyneuropathie, atteinte endocrinienne, organomégalies et signes cutanés) et du syndrome de Schnitzler (urticaire, fièvre, arthralgies et ostéocondensations) ;
– l’immunoglobuline monoclonale peut précipiter (cryoglobulinémies) ;
– l’immunoglobuline monoclonale peut se déposer dans n’importe quel organe. L’amylose AL est ainsi diagnostiquée sur une augmentation de chaîne légère lambda ou sur une biopsie d’organes (glandes salivaires accessoires). Elle peut compliquer un myélome connu ou être « primitive ». Ses dépôts atteignent particulièrement le rein, avec une protéinurie souvent abondante, le cœur, les nerfs périphériques, le système digestif et les tissus mous (macroglossie, syndrome du canal carpien, hématomes palpébraux, etc.).
Découverte fortuite d’un « micro-pic »
Cette situation n’est pas exceptionnelle, en particulier chez le sujet âgé. La première chose à faire est l’EPS à trois mois pour éliminer un pic transitoire (concomitant d’une maladie infectieuse, par exemple). S’il est confirmé, on peut surseoir au myélogramme dans les conditions suivantes :
– patient de plus de 60 ans ;
– dysglobulinémie de type IgG ;
– pic < 15 g/L ;
– critères CRAB négatifs (
– ratio chaînes légères sériques normal.
Il faut cependant surveiller le patient à six mois, puis annuellement, par un examen clinique et un contrôle de l’EPS, de la calcémie, de la créatininémie et de l’hémogramme. Dans toute autre situation, le recours à un avis spécialisé s’impose.
1. Électrophorèse des protides sériques normale (fig. 1)
• Albumine : 40 à 45 g/L
• Alpha-1-globulines : 2 à 4 g/L
• Alpha-2-globulines : 4,5 à 7,5 g/L
• Bêtaglobulines : 7 à 13 g/L
• Gammaglobulines : 5 à 15 g/L
• Protides totaux : 50 à 95 g/L
2. Critères diagnostiques des MGUS
Dysglobulinémie monoclonale non IgM (IgG, IgA, IgD) :
– protéines monoclonales < 30 g/L ;
– plasmocytose médullaire au myélogramme < 10 % ;
– pas d’atteinte d’organes (critères CRAB négatifs ; tableau 2).
Dysglobulinémie monoclonale IgM :
– protéines monoclonales < 30 g/L ;
– lymphocytose médullaire < 10 % ;
– pas d’atteinte d’organes (anémie, adénopathies, hépatosplénomégalie, manifestations d’hyperviscosité).
Que dire à vos patients ?
L’hypogammaglobulinémie justifie un bilan simple. Ce n’est pas le taux de gammaglobulines qui fait prescrire un traitement substitutif mais la fréquence des infections ORL ou pulmonaires.
La découverte d’une hypergammaglobulinémie polyclonale nécessite d’en rechercher la cause, mais elle n’est pas pathogène en elle-même.
La MGUS a un risque relativement faible d’évoluer vers une maladie tumorale mais justifie une surveillance régulière et prolongée par des examens simples.
En cas de suspicion de myélome, de maladie de Waldenström ou de maladie systémique associée, une prise en charge spécialisée est nécessaire.
1. HAS. Quand prescrire une électrophorèse des protéines sériques (EPS) et conduite à tenir en cas de découverte d’une immunoglobuline monoclonale. Recommandation de bonne pratique. 30 janvier 2017.
2. Samson M, Audia S, Lakomy D, et al. Stratégie diagnostique devant une hypogammaglobulinémie en rhumatologie. Rev Rhum 2011;78:122-7.
3. Fieschi C, Viallard JF. Les déficits immunitaires communs variables (DICV). Partie 1 : évolution des critères diagnostiques et des connaissances génétiques. Rev Med Int 2021;42(7):465-72.
4. Zhao EJ, Cheng CV, Mattman A, et al. Polyclonal hypergammaglobulinaemia: assessment, clinical interpretation and management. Lancet Haematol 2021;8(5):e365-75.
5. Beuvon C, Puyade M, Martellosio JP, et al. Étiologies associées aux hypergammaglobulinémies polyclonales : revue systématique de la littérature. Rev Med Int 2017;38(1):133-4.
6. Therneau TM, Kyle RA, Melton LJ, et al. Incidence of monoclonal gammapathy of undetermined significance and estimation of duration before first clinical recognition. Mayo Clin Proc 2012;87(11):1071-9.
7. Wadhera RK, Rajkumar SV. Prevalence of monoclonal gammopathy of undetermined significance: a systematic review. Mayo Clin Proc 2010;85(10):933-42.
8. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014;15(12):e538-48.
9. Dejoie T, Lakomy D, Caillon H, et al. Recommandations de l’Intergroupe francophone du myélome pour l’harmonisation de l’analyse des électrophorèses des protéines sériques et urinaires dans le diagnostic et le suivi du myélome multiple. Hématologie 2017;23:312-24.
Dans cet article
 Encadrés
Encadrés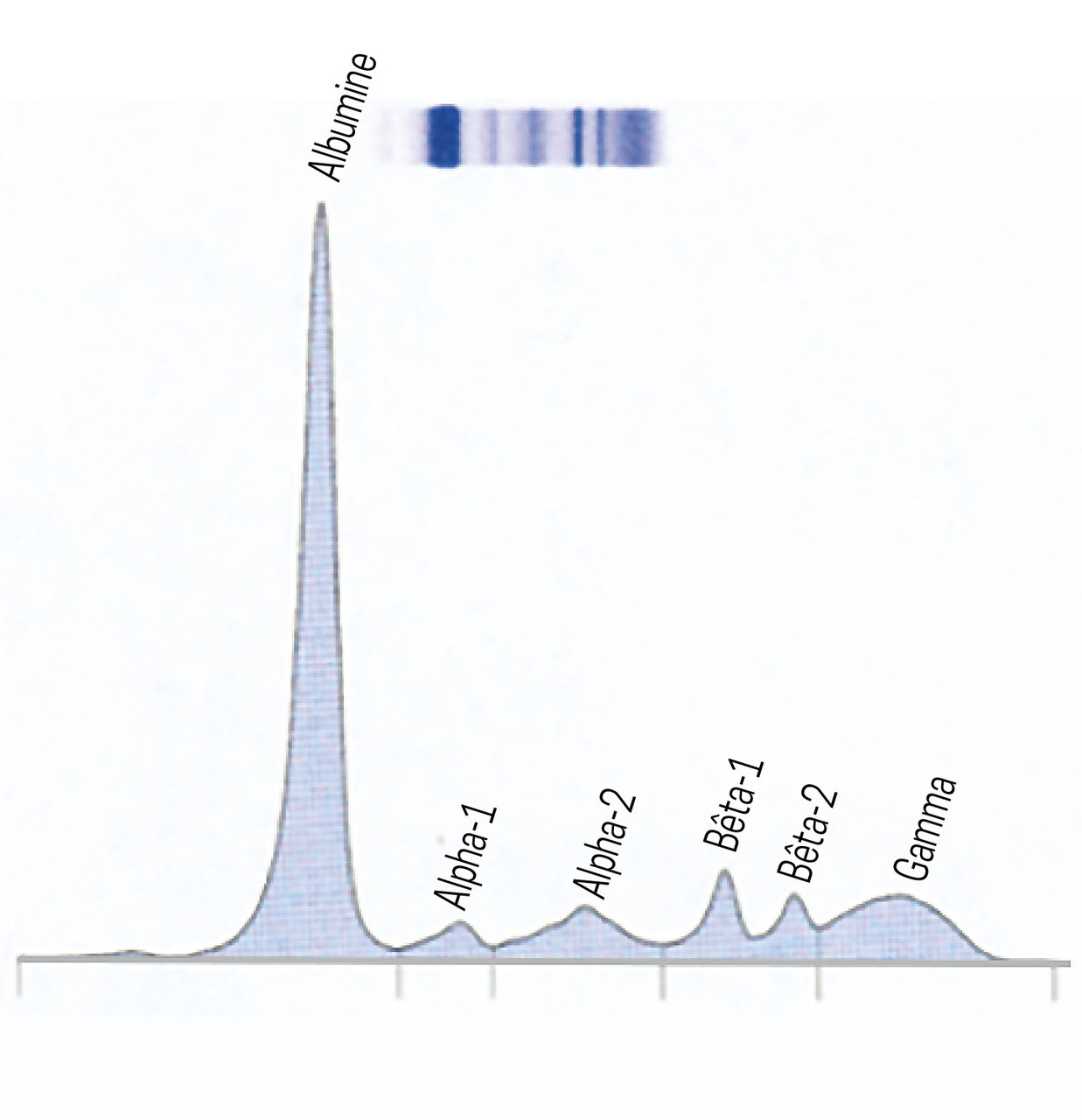




L’EPS doit être prescrite selon les recommandations de 2017.
Les hypogammaglobulinémies doivent faire évoquer un DICV chez le sujet jeune et un myélome à chaîne légère ou une pathologie lymphoïde chez le patient de plus de 60 ans.
Les hypergammaglobulinémies polyclonales orientent principalement vers les hépatopathies chroniques, certaines maladies systémiques, le VIH, les lymphomes.
La découverte d’un pic monoclonal impose un typage par immunofixation. Après élimination d’un pic transitoire, il importe de savoir si l’on se trouve dans une situation prétumorale ou tumorale.