La maladie de Behçet est une vascularite systémique, touchant les petits et grands vaisseaux.1 Elle atteint préférentiellement les sujets d’âge jeune (de 10 à 45 ans). Une première poussée après 50 ans est rare. Les formes infantiles sont de plus en plus fréquemment rapportées. La maladie de Behçet concerne autant les hommes que les femmes. Les formes symptomatiques et notamment sévères sont toutefois plus fréquentes chez l’homme. La grossesse ne semble pas avoir d’influence sur cette maladie (pas d’augmentation du risque de poussée). Cependant, de très rares cas néonataux ont été décrits, avec une atteinte cutanée le plus souvent transitoire.
La maladie de Behçet est ubiquitaire mais plus fréquente dans les populations issues des pays situés sur la route de la soie (de l’est de l’Asie au Bassin méditerranéen).
Son incidence en France est de 7/100 000, et un antécédent familial augmente la probabilité diagnostique.
Il n’existe aucun marqueur d’activité biologique pour cette maladie. Le diagnostic repose donc sur un faisceau d’arguments cliniques et se définit par un score supérieur ou égal à 4 selon les critères de classification internationaux de la maladie de Behçet révisés en 2013 (
Perturbation de l’homéostasie des réponses immunitaires innée et adaptative ?
La physiopathologie de la maladie de Behçet n’est pas complètement comprise à l’heure actuelle. L’hypothèse la plus fréquente, mais jamais démontrée, est celle d’une réponse immunitaire anormale secondaire à une infection (streptocoque, virus Herpes simplex). De nombreux effecteurs immunitaires sont mis en jeu : lymphocytes T auxiliaires Th1 et gamma/delta, polynucléaires neutrophiles et lymphocytes natural killer (NK). Une étude récente a mis en évidence des perturbations de l’homéostasie lymphocytaire (augmentation des lymphocytes T auxiliaires Th17 et diminution des lymphocytes T régulateurs) impliquant différentes cytokines (interleukines 17, 23 et 21).3 Ces réactions inflammatoires anormales surviendraient chez des sujets génétiquement prédisposés : le risque de développer la maladie est multiplié par 5 chez les patients porteurs de l’haplotype HLA-B51, et de nouveaux gènes de susceptibilité (IL-10, IL-23R, IL-12Rβ2) ont été récemment retrouvés.
Un tableau clinique polymorphe
Atteinte cutanéomuqueuse, la plus fréquente
L’aphte buccal, quasi constant, est le symptôme le plus fréquent (
Les aphtes génitaux (présents dans 50 à 70 % des cas) sont très évocateurs de la maladie de Behçet. Chez l’homme, ils siègent sur le scrotum, plus rarement sur la verge ou l’urètre. Chez la femme, la vulve, le vagin et le col peuvent être atteints. Disséminés et douloureux, ou totalement latents, les aphtes génitaux sont souvent plus profonds et de plus grande taille que les aphtes buccaux. Ils récidivent moins que ces derniers mais laissent des cicatrices dépigmentées permettant un diagnostic rétrospectif.
Les aphtes peuvent également siéger au niveau de la muqueuse digestive (œsophage, estomac, intestin).
Les atteintes cutanées, également fréquentes (70 % des patients), peuvent prendre la forme d’un érythème noueux, de pseudofolliculite (pustule non centrée sur un follicule pileux), de nodules acnéiformes, de thrombophlébite superficielle migratoire, d’hyperréactivité cutanée aspécifique aux agressions de l’épithélium, de purpura, de lésions nécrotiques par vascularite cutanée et de lésions de dermatose neutrophilique. L’érythème noueux touche un tiers des patients. Il se manifeste par des nodules douloureux, érythémateux, dont l’évolution suit classiquement toutes les phases de la biligénie. Ils ne doivent pas être confondus avec les thromboses superficielles, qui peuvent avoir une présentation clinique assez proche. L’hypersensibilité cutanée après un traumatisme est à l’origine du test pathergique, considéré comme positif lorsque qu’une papule/pustule apparaît dans les 24 à 48 heures après la piqûre de l’avant-bras par une aiguille 21G (8/10 mm). La sensibilité de ce test est moins bonne en cas d’usage de dispositifs jetables ou de désinfection locale.
Atteinte oculaire, à haut risque de séquelles graves
L’atteinte oculaire est fréquente et particulièrement sévère.4 Elle conditionne le pronostic fonctionnel des patients avec un risque élevé de cécité (10-15 % sous traitement immunosuppresseur adapté et 50 % sans traitement), d’autant que les formes oculaires sont volontiers bilatérales (70 % à 2 ans). L’examen ophtalmologique doit être systématique car les lésions peuvent être initialement paucisymptomatiques.
L’inflammation intraoculaire évolue par poussées et concerne les chambres antérieure et/ou postérieure. Les atteintes les plus fréquemment observées sont les uvéites postérieures ou les panuvéites. Les uvéites postérieures sont souvent sévères, avec un œdème maculaire responsable d’une baisse de l’acuité visuelle importante. On observe également des vascularites rétiniennes occlusives et nécrosantes. Les uvéites antérieures, le plus souvent associées à une atteinte du segment postérieur (90 % des cas), non granulomateuses, exposent aux synéchies cristalliniennes et à l’hypertonie oculaire par défaut de résorption de l’humeur aqueuse. Elles peuvent se compliquer d’hypopion, très évocateur de la maladie de Behçet (peu de diagnostics différentiels hormis les maladies inflammatoires chroniques intestinales et l’uvéite herpétique). Aphte conjonctival, épisclérite et kératite ont également été décrits mais sont plus rares.
Atteinte articulaire : 1 patient sur 2
L’atteinte articulaire est précoce, parfois inaugurale, pouvant précéder de plusieurs années les autres manifestations. Le plus souvent, il s’agit d’arthralgies, voire d’oligoarthrites fixes, siégeant sur les articulations porteuses (genoux, chevilles). L’évolution est récidivante et asymétrique. Les formes polyarticulaires sont rares (2 %). Les radiographies sont normales et les destructions articulaires sont exceptionnelles. La ponction articulaire met le plus souvent en évidence un liquide inflammatoire riche en polynucléaires neutrophiles.
Une myosite vraie peut parfois s’associer aux atteintes articulaires. Elle est alors focale, localisée aux membres inférieurs et plus rarement orbitaire. Le patient se plaint de myalgies. Les enzymes musculaires (créatine phosphokinases [CPK]) sont exceptionnellement élevées. Une rhabdomyolyse secondaire à la colchicine ou une myopathie doivent être évoquées si le dosage des CPK est élevé. La thombophlébite est le diagnostic différentiel à éliminer dans les formes localisées. L’imagerie par résonance magnétique (IRM) musculaire est indiquée, et la biopsie musculaire peut objectiver une vascularite focale.
Des lésions neurologiques, facteur de gravité
Les atteintes neurologiques sont observées dans 20 % des cas en moyenne (4 à 42 % selon les séries). Elles peuvent prendre différentes formes : méningite ou méningo-encéphalite, atteinte du parenchyme cérébral, thrombose veineuse centrale, vascularite des artères à destination cérébrale. Elles entraînent des séquelles fonctionnelles qui en font un des facteurs de gravité de la maladie.5 Parfois précédés de fièvre, les signes neurologiques possibles sont nombreux : céphalées, syndrome pyramidal, ataxie cérébelleuse, troubles sphinctériens, paralysie des nerfs crâniens, voire ralentissement psychomoteur et troubles du comportement. La ponction lombaire met en évidence une méningite lymphocytaire, parfois puriforme. L’imagerie par résonance magnétique peut montrer des hypersignaux diffus, très évocateurs quand ils atteignent le tronc cérébral. Dans certaines séries, 20 % des patients ont un handicap à quatre ans. La mortalité est en grande partie liée aux complications de décubitus. Le pronostic, encore sévère, est cependant amélioré par l’association thérapeutique de corticoïdes et d’immunosuppresseurs, surtout quand ils sont instaurés précocement.
Atteintes vasculaires : très évocatrices
Les atteintes vasculaires sont plus fréquentes chez les sujets jeunes et de sexe masculin.
L’atteinte veineuse survient en général au début de la maladie et concerne 14 à 39 % des patients, selon les séries.6 Si les thromboses veineuses superficielles et profondes des membres inférieurs sont les plus fréquentes (52 %), des atteintes plus graves des gros troncs veineux sont souvent décrites (30 % des patients environ) et font la particularité de cette maladie. 20 % des patients ont déjà eu une complication veineuse au moment où le diagnostic est posé. Un tableau fébrile et un syndrome inflammatoire biologique sont volontiers présents au diagnostic de l’atteinte vasculaire. On retrouve, selon les séries, une thrombose des veines cérébrales chez plus de 10 % des patients, une embolie pulmonaire dans 2 à 27 % des cas ; la fréquence de la thrombose de la veine cave inférieure est de plus de 7 % et celle de la thrombose de la veine cave supérieure est de 3 %. Le syndrome de Budd-Chiari est également noté dans 3 % des cas. Plus rarement, on peut observer une thrombose cardiaque (oreillette et ventricule droit), une thrombose rénale, portale ou rétinienne. Les thrombophlébites superficielles font partie des complications veineuses les plus fréquentes, avec 2 à 47 % des patients concernés selon les séries. Elles prennent la forme de nodules érythémateux sensibles à disposition parfois linéaire, souvent confondues avec un érythème noueux.
L’atteinte artérielle touche 4 à 17 % des patients selon les séries. Elle est probablement sous-estimée puisqu’une série autopsique japonaise a retrouvé une atteinte artérielle chez 34 % de 170 patients.7 L’atteinte artérielle de la maladie de Behçet concerne des vaisseaux de différents calibres. Elle survient précocement dans l’histoire naturelle de la maladie (symptôme initial chez 8 % des patients). Un « traumatisme artériel » est retrouvé chez environ 20 % des patients (chirurgie, artériographie, gaz du sang, etc.). Les principales atteintes artérielles retrouvées sont les suivantes : anévrismes (45-70 %), occlusions (36-80 %) et plus rarement sténoses artérielles (13 %) ou aortites (3 %). Les principales localisations sont aortiques (abdominale dans 11 % des cas et thoracique dans 5 % des cas), fémorale (15 %), pulmonaire (14 %) et iliaque (14 %). Les atteintes artérielles représentent la principale cause de décès (entre 30 et 40 %).
Des vascularites des petits vaisseaux sont également observées, avec des anomalies à la capillaroscopie chez 50 à 75 % des patients ayant une maladie de Behçet. Les lésions sont de plusieurs types : dystrophies capillaires, nombre anormal de capillaires, pétéchies ou œdème péricapillaire.
Atteinte cardiaque : les trois tuniques
Cette atteinte est particulièrement rare puisqu’elle ne concerne que 5 % des patients environ. Les complications cardiaques peuvent toucher les trois tuniques (myocarde, endocarde et péricarde). L’atteinte coronarienne est aussi possible (anévrisme et thromboses compliquées d’infarctus du myocarde).
Atteintes gastro-intestinales : des lésions peu spécifiques
Les lésions digestives macroscopiques de la maladie de Behçet ressemblent à celles de la rectocolite hémorragique et de la maladie de Crohn. Leur fréquence est très variable selon les séries (de 5 à 30 %). La symptomatologie et les aspects endoscopique et histologique sont aspécifiques. Cependant, contrairement à la maladie de Crohn, il n’y a jamais de granulome sur les biopsies.
Les autres organes cibles
L’atteinte pulmonaire est très rare. Elle se caractérise essentiellement par des lésions nodulaires de consolidation post-infarctus pulmonaire qui ont tendance à s’excaver, avec ou sans pleurésie associée. On peut parfois observer des infiltrats en verre dépoli, stigmates d’une hémorragie intra-alvéolaire. Une vascularite est aussi possible dans cette localisation.
Les atteintes rénales sont exceptionnelles et dominées par la néphropathie amyloïde, qui survient chez des patients non contrôlés après plusieurs années d’évolution.
Enfin, l’atteinte testiculaire et épididymaire (orchiépididymite) est aussi rapportée.
Des diagnostics différentiels variables selon le tableau clinique
Devant une aphtose buccale et/ou génitale récidivante, plusieurs autres hypothèses diagnostiques se discutent : l’aphtose buccale bénigne récidivante idiopathique, les maladies inflammatoires chroniques intestinales (MICI), les syndromes auto-inflammatoires mendéliens (haplo-insuffisance d’A20, déficit en mévalonate kinase) et les « Behçet-like » syndromiques. Lorsque le patient a des ulcérations buccales non identifiées, il convient de prendre l’avis d’un dermatologue pour éliminer notamment des infections herpétiques (lésions érosives et vésiculaires) ou des maladies bulleuses avec atteinte de la cavité buccale (pemphigoïde, pemphigus vulgaire, pemphigus superficiel nécessitant une biopsie). Toute ulcération muqueuse peut aussi être iatrogénique, secondaire à une neutropénie ou à une carence vitaminique.
Les symptômes neurologiques peuvent rappeler ceux de la sclérose en plaques, de la sarcoïdose, de lésions tumorales, d’un lymphome ou d’une méningo-encéphalite infectieuse.
Les atteintes articulaires peuvent évoquer des spondylarthropathies, qui doivent aussi être recherchées et écartées.
Enfin, les diagnostics différentiels des manifestations digestives à évoquer, outre les MICI, sont les colites infectieuses et la toxicité éventuelle d’anti-inflammatoires non stéroïdiens (AINS).
Principes du traitement de la maladie de Behçet
Le pronostic de la maladie de Behçet est péjoratif en cas de lésions artérielles ou cardiaques (mortalité accrue) et en cas d’atteinte oculaire et/ou neurologique (morbidités majeures).9 Le choix du traitement dépend des atteintes cliniques (
Prise en charge de la forme cutanéomuqueuse ou articulaire
La colchicine est le traitement de première intention (en l’absence de contre-indication) pour prévenir la récidive des lésions cutanéomuqueuses, à une posologie comprise entre 1 et 2 mg/j. Elle est bien tolérée, en dehors de troubles digestifs (diarrhées), qui cèdent en règle spontanément après quelques jours ou peuvent être atténués en y associant tiémonium et opiacés. La toxicité de la colchicine peut être accrue par la prise de médicaments interférant avec le métabolisme du cytochrome P450 (pyostacine ou macrolides, par exemple). Son utilisation pendant la grossesse est possible. En parallèle, une bonne hygiène buccodentaire est essentielle. Les dermocorticoïdes de classe I ou II peuvent être proposés pour le traitement de l’aphtose buccale et génitale. En cas de lésions buccales invalidantes, les bains de bouche avec des corticoïdes peuvent être utiles en complément. La xylocaïne en gel peut soulager les ulcères génitaux très douloureux. Chez les patients réfractaires avec une atteinte muqueuse sévère, l’aprémilast (inhibiteur de la phosphodiestérase 4) a obtenu une autorisation de mise sur le marché dans cette indication.10 Il est prescrit à la dose de 30 mg × 2/j. Des troubles digestifs (diarrhées, nausées) sont observés dans 30-40 % des cas dans les premières semaines de traitement et sont atténués par la prescription de traitements symptomatiques. La thalidomide est une alternative intéressante, mais les risques tératogènes, de thrombose et de neuropathie limitent sont utilisation. D’autres traitements tels que les anti-TNFα, l’azathioprine ou l’ustékinumab peuvent être envisagés dans les formes réfractaires.
Comme la prévention des récidives cutanéomuqueuses, l’atteinte articulaire relève d’un traitement de première intention par la colchicine (1 à 2 mg/j). Les infiltrations de corticoïdes peuvent y être associées. En traitement des poussées, les AINS ou des cures courtes de prednisone orale sont proposés. Dans les formes réfractaires ou récidivantes, un traitement de fond devient nécessaire : méthotrexate (0,3 mg/m²/sem) ou anti-TNFα.
Traitement de l’atteinte oculaire, vasculaire sévère ou neurologique : éviter les séquelles
Lorsqu’elle touche le segment postérieur, l’atteinte oculaire justifie systématiquement l’emploi d’immunosuppresseurs en plus de la corticothérapie (0,5 à 1 mg/kg/j, plus ou moins bolus de méthylprednisolone à la dose de 500 mg/j pendant 3 jours). L’azathioprine (2,5 mg/kg/j) est ainsi utilisée en l’absence de signes de gravité ; les anti-TNFα ou l’interféron α sont recommandés dans les formes sévères.
Les recommandations nationales (Protocole national de diagnostic et de soins [PNDS])11 et internationales12 préconisent de traiter les thromboses veineuses profondes et les anévrismes artériels par immunosuppresseurs : azathioprine pour les thromboses veineuses des membres et les anévrismes périphériques ; cyclophosphamide ou anti-TNFα dans les atteintes plus sévères (syndrome de Budd-Chiari, thrombose de la veine cave inférieure, anévrisme de l’aorte ou des artères pulmonaires). Le rôle favorable des anticoagulants au cours des complications veineuses reste débattu. Cependant, le PNDS de la maladie de Behçet recommande l’anticoagulation dans les thromboses veineuses en l’absence de lésions artérielles anévrismales à risque hémorragique.
L’atteinte neurologique, enfin, justifie l’emploi de l’azathioprine en plus de la corticothérapie. Bolus de Solumedrol, cyclophosphamide ou anti-TNF α sont prescrits dans les formes sévères.
L’haplo-insuffisance d’A20, un diagnostic différentiel de la maladie de Behçet à connaître
Cette maladie autosomique dominante rare est liée à une mutation du gène TNFAIP3 (tumor necrosis factor α-induced protein 3, A20) se situant sur le chromosome 6. Le début des symptômes est plus précoce que dans la maladie de Behçet (avant 10 ans). Elle se manifeste par une aphtose bipolaire sévère et parfois délabrante associée à une uvéite, antérieure et isolée dans la majorité des cas.8 Les épisodes fébriles sont plus constants que dans la maladie de Behçet. Les poussées inflammatoires s’accompagnent d’une élévation nette de la protéine C-réactive (CRP). Les patients peuvent avoir au cours de leur vie des manifestations auto-immunes et parfois un déficit immunitaire humoral discret.
1. Saadoun D, Vautier M, Cacoub P. Medium –and Large– Vessel Vasculitis. Circulation 2021;143(3):267-82.
2. The International Criteria for Behcet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol 2014;28(3):338-47.
3. Geri G, Terrier B, Rosenzwajg M, et al. Critical role of IL-21 in modulating TH17 and regulatory T cells in Behcet disease. J Allergy Clin Immunol 2011;128(3):655-64.
4. Saadoun D, Cassoux N, Wechsler B, et al. Ocular manifestations of Behcet’s disease. Rev Med Interne 2010;31(8):545-50.
5. Noel N, Bernard R, Wechsler B, et al. Long-term outcome of neuro-Behçet’s disease. Arthritis Rheumatol 2014;66(5):1306-14.
6. Desbois AC, Wechsler B, Resche-Rigon M, et al. Immunosuppressants reduce venous thrombosis relapse in Behcet’s disease. Arthritis Rheum 2012;64(8): 2753-60.
7. Lakhanpal S, Tani K, Lie JT, et al. Pathologic features of Behçet’s syndrome: a review of Japanese autopsy registry data. Hum Pathol 1985;16(8):790-5.
8. Kone-Paut I, Barete S, Bodaghi B, et al. French recommendations for the management of Behcet’s disease. Orphanet J Rare Dis 2021;16:352.
9. Saadoun D, Wechsler B, Desseaux K, et al. Mortality in Behcet’s disease. Arthritis Rheum 2010;62(9): 2806-12.
10. Hatemi G, Mahr A, Ishigatsubo Y, et al. Trial of Apremilast for Oral Ulcers in Behcet’s Syndrome. N Engl J Med 2019;381(20):1918-28.
11. Saadoun D. Behçet’s disease: The French recommendations. Rev Med Interne 2020;41(7):437-9.
12. Hatemi G, Christensen R, Bang D, et al. 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis 2018;77(6): 808-18.
Dans cet article
 Encadrés
Encadrés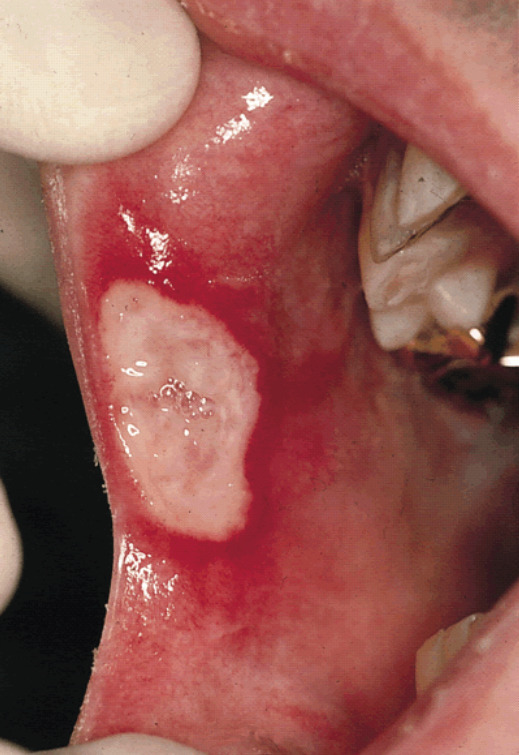
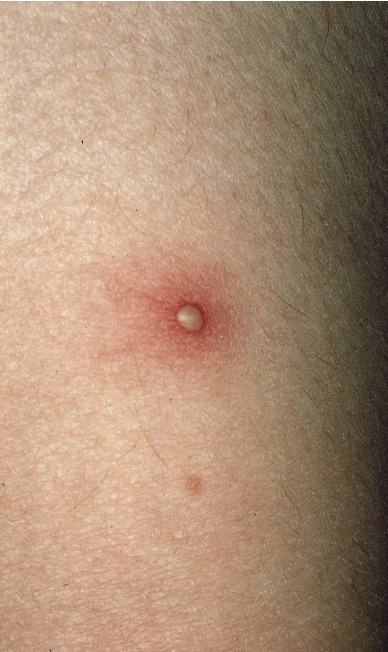
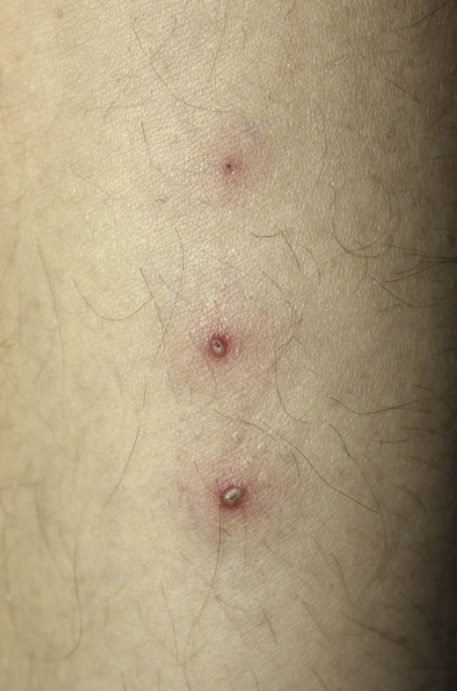

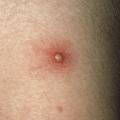
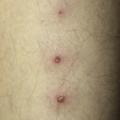
Le diagnostic de la maladie de Behçet, vascularite systémique des sujets jeunes, est clinique.
Les ulcérations cutanéomuqueuses sont les manifestations les plus fréquentes.
L’évolution se fait par poussées, avec des périodes d’accalmie.
Le pronostic est lié aux atteintes artérielle (augmentation de la mortalité), oculaire et neurologique (séquelles graves).