Le prix Nobel de physiologie et de médecine a été attribué cette année à Gregg L. Semenza (Baltimore), sir Peter J. Ratcliffe (Oxford) et William G. Kaelin Jr (Harvard) pour leurs travaux de recherche sur les mécanismes moléculaires impliqués dans l’adaptation des cellules aux variations du taux d’oxygène.
Description de la voie de l’hypoxie et des découvertes réalisées
Ces recherches ont eu comme point de départ l’identification, en 1990, des mécanismes responsables de la régulation de l’érythropoïétine (EPO), hormone qui stimule la production de globules rouges en réponse à la baisse du taux d’oxygène (hypoxie).
Érythropoïétine
Dès 1863, une association entre la survenue de polyglobulies et la vie en haute altitude avait été notée, mais c’est en 1905 que Paul Carnot identifie l’EPO (appelée initialement hémopoïétine) après avoir constaté que l’injection de sérum de lapins anémiques stimulait la production de globules rouges chez des lapins normaux. La synthèse de l’EPO par le rein a été découverte en 1957, puis il a été montré qu’elle était également produite par le foie fœtal. Le gène codant l’EPO a été découvert en 1985, et le principal site de production de l’EPO chez l’adulte a été identifié en 1988 dans les cellules interstitielles péritubulaires du rein.
Un facteur nommé HIF
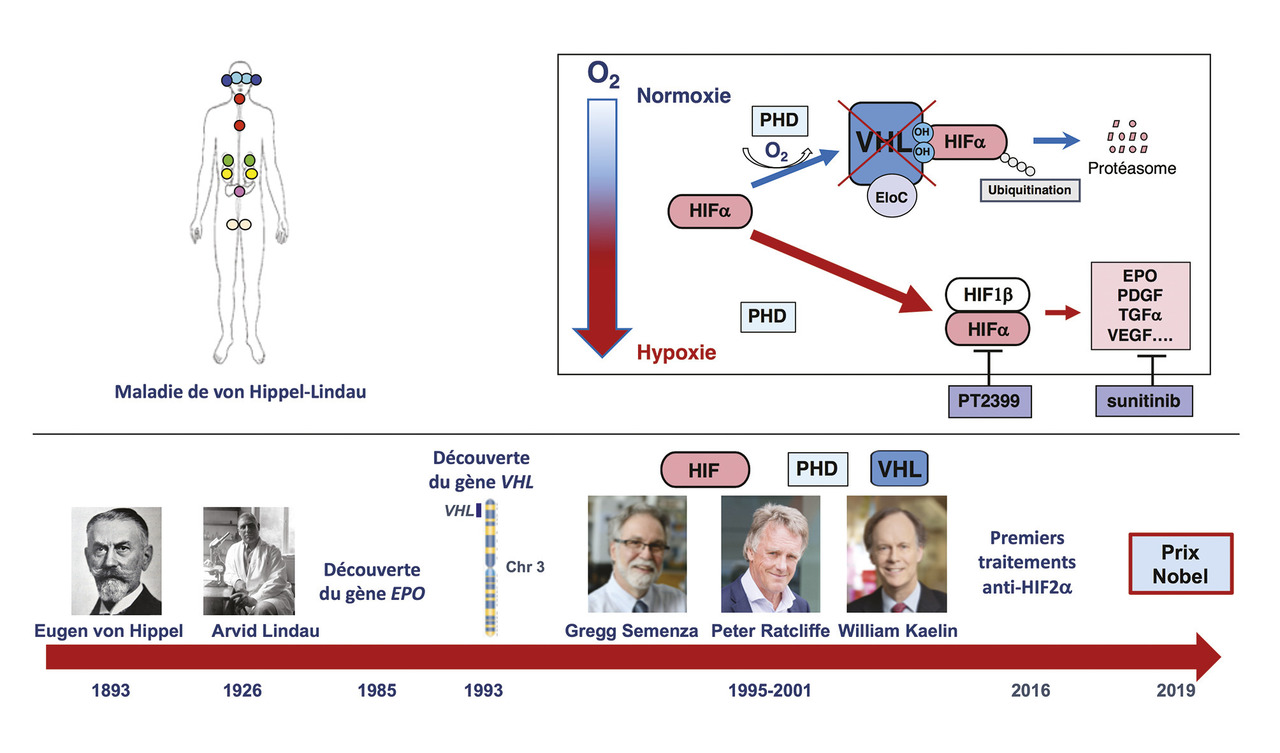
C’est sur la base de ces travaux que Gregg Semenza puis Peter Ratcliffe (fig. 1 ) ont débuté leurs recherches. L’utilisation de lignées de souris transgéniques dans lesquelles le gène de l’EPO humaine avait été inséré a permis la mise en évidence d’une région capable de stimuler son expression par l’hypoxie et de fixer un facteur appelé hypoxia inducible factor (HIF) identifié en 1992 et purifié en 1995 par Guo-Liang Wang et Gregg Semenza.1 Il s’agit d’un hétérodimère constitué d’une sous-unité alpha (HIF1, HIF2 ou HIF3) et d’une sous-unité bêta.
En 1998, Semenza a montré que la perte de HIF1 alpha était létale chez la souris et que HIF1 alpha jouait également un rôle majeur dans le métabolisme glycolytique, confirmant des résultats précédemment obtenus par Ratcliffe. La même année, l’implication de HIF dans l’angiogenèse tumorale, la prolifération cellulaire et l’apoptose était aussi démontrée.
La communauté scientifique et médicale s’est très vite intéressée à cette nouvelle voie biologique, notamment dans le domaine du cancer, dont une des caractéristiques est la diminution du taux d’oxygène dans les tumeurs en prolifération, à l’origine de la formation de néovaisseaux sanguins (angiogenèse tumorale).
En 1998, Semenza a montré que la perte de HIF1 alpha était létale chez la souris et que HIF1 alpha jouait également un rôle majeur dans le métabolisme glycolytique, confirmant des résultats précédemment obtenus par Ratcliffe. La même année, l’implication de HIF dans l’angiogenèse tumorale, la prolifération cellulaire et l’apoptose était aussi démontrée.
La communauté scientifique et médicale s’est très vite intéressée à cette nouvelle voie biologique, notamment dans le domaine du cancer, dont une des caractéristiques est la diminution du taux d’oxygène dans les tumeurs en prolifération, à l’origine de la formation de néovaisseaux sanguins (angiogenèse tumorale).
Le gène VHL
C’est ainsi que le Pr William Kaelin (fig. 1 ), spécialiste de la maladie de von Hippel-Lindau (VHL), a rapidement consacré ses recherches à la voie de l’hypoxie. En effet, cette prédisposition héréditaire rare (1/36 000 naissances) est caractérisée par le développement de tumeurs richement vascularisées dans le système nerveux central et la rétine (hémangioblastomes, tumeurs du sac endolymphatique), les reins (carcinomes à cellules claires), le pancréas (tumeurs endocrines) et les surrénales (phéochromocytomes) [v. le site du réseau PREDIR : http.predir.org).2 Le gène VHL, dont les mutations constitutionnelles sont responsables de l’affection, a été identifié en 1993. Dès 1995, l’équipe de Kaelin a mis en évidence le rôle suppresseur de tumeur du gène VHL dans les cancers du rein et montré que celui-ci était lié à l’inhibition des gènes de la voie de l’hypoxie.3 En 1999, l’équipe de Ratcliffe a identifié le rôle de VHL dans l’ubiquitination et la dégradation de HIF de façon dépendante de l’oxygène.4
Le lien entre VHL, HIF et l’oxygène
Le lien final entre VHL, HIF et l’oxygène a été élucidé en 2001, lorsque les groupes de Ratcliffe5 et Kaelin6 ont publié dans le même numéro de Science leurs travaux montrant qu’une réaction d’hydroxylation de HIF1alpha dépendante de l’oxygène permettait sa fixation à la protéine VHL, son ubiquitination, puis sa dégradation dans le protéasome. Cette fixation n’est plus possible en cas de mutations du gène VHL, ce qui entraîne l’expression des gènes cibles de HIF et conduit au développement de tumeurs richement vascularisées.
Parallèlement, Ratcliffe a identifié les enzymes responsables de l’hydroxylation de HIF (prolyl hydroxylase domain protein [PHD]) dont l’activité est dépendante de la présence d’oxygène, de fer et d’acide ascorbique ainsi que du 2-oxoglutarate (2-OG).7 Ces enzymes sont ainsi les véritables senseurs d’oxygène de la voie de régulation de l’hypoxie (fig. 2 ).
Ces réactions d’hydroxylation sont très sensibles aux métabolites comme le fumarate et le succinate qui s’accumulent lors de mutations des gènes codant la succinate déshydrogénase (SDH) ou la fumarate hydratase (FH) observées dans certaines tumeurs sporadiques et héréditaires (paragangliomes, phéochromocytomes, cancers rénaux). L’inhibition des hydroxylases a alors pour conséquence l’activation de la voie de l’hypoxie qui participe à la tumorigenèse. Plus récemment, il a été montré que les mutations dans les gènes IDH1 et IDH2 dans les gliomes entraînent également l’inhibition des PHD et participent ainsi à l’angiogenèse de ces tumeurs.
Parallèlement, Ratcliffe a identifié les enzymes responsables de l’hydroxylation de HIF (prolyl hydroxylase domain protein [PHD]) dont l’activité est dépendante de la présence d’oxygène, de fer et d’acide ascorbique ainsi que du 2-oxoglutarate (2-OG).7 Ces enzymes sont ainsi les véritables senseurs d’oxygène de la voie de régulation de l’hypoxie (
Ces réactions d’hydroxylation sont très sensibles aux métabolites comme le fumarate et le succinate qui s’accumulent lors de mutations des gènes codant la succinate déshydrogénase (SDH) ou la fumarate hydratase (FH) observées dans certaines tumeurs sporadiques et héréditaires (paragangliomes, phéochromocytomes, cancers rénaux). L’inhibition des hydroxylases a alors pour conséquence l’activation de la voie de l’hypoxie qui participe à la tumorigenèse. Plus récemment, il a été montré que les mutations dans les gènes IDH1 et IDH2 dans les gliomes entraînent également l’inhibition des PHD et participent ainsi à l’angiogenèse de ces tumeurs.
Applications en médecine
La voie de l’hypoxie est dérégulée dans de nombreuses pathologies (anémies, cancers, maladies ischémiques et inflammatoires)8 et, rapidement, de nombreuses stratégies thérapeutiques ont été explorées et développées pour pallier ces altérations.9
Traitement contre le cancer
Les études du gène suppresseur de tumeur VHL ont été le point de départ du développement de thérapies anti- angiogéniques. En effet, il a été découvert que celui-ci était muté dans 70 % des carcinomes rénaux à cellules claires qui ont les taux de vascular endothalial growth factor (VEGF) [gène cible majeur de HIF] les plus élevés des tumeurs solides. Les cancers du rein se sont ainsi révélés très sensibles aux traitements anti- angiogéniques ciblant initialement le VEGF (bévacizumab) puis son récepteur kinase (sunitinib, sorafénib, axitininib, pazopanib, cabozantinib).
Plus récemment ont été mis au point des inhibiteurs de HIF2 alpha10, et les premiers essais cliniques réalisés avec l’un d’entre eux (PT-2385) chez des patients atteints de carcinome rénal à cellules claires métastatiques sont encourageants.11
L’utilisation de ces composés est également envisagée dans le traitement de l’hypertension artérielle pulmonaire (HTAP), notamment développée chez les patients polyglobuliques porteurs de mutations germinales dans les gènes codant HIF2 alpha ou VHL. Les premiers cas décrits concernaient une population de l’Europe de l’Est (république de Tchouvachie), porteuse d’une mutation particulière sur les deux allèles du gène VHL (Arg200Trp). Ces patients ont une polyglobulie sévère associée à des accidents thrombotiques, de l’HTAP et des angiomes vertébraux. D’autres mutations du gène VHL puis des mutations dans les gènes PHD1, PHD2 et HIF2A ont ensuite été identifiées chez des patients atteints de polyglobulie congénitale. Les mutations du gène VHL responsables de polyglobulies sont moins délétères que celles décrites chez les patients avec maladie de VHL. Ces mutations entraînent en effet une faible activation de la voie de l’hypoxie, ce qui expliquerait l’absence de développement tumoral chez les patients polyglobuliques.12
Plus récemment ont été mis au point des inhibiteurs de HIF2 alpha10, et les premiers essais cliniques réalisés avec l’un d’entre eux (PT-2385) chez des patients atteints de carcinome rénal à cellules claires métastatiques sont encourageants.11
L’utilisation de ces composés est également envisagée dans le traitement de l’hypertension artérielle pulmonaire (HTAP), notamment développée chez les patients polyglobuliques porteurs de mutations germinales dans les gènes codant HIF2 alpha ou VHL. Les premiers cas décrits concernaient une population de l’Europe de l’Est (république de Tchouvachie), porteuse d’une mutation particulière sur les deux allèles du gène VHL (Arg200Trp). Ces patients ont une polyglobulie sévère associée à des accidents thrombotiques, de l’HTAP et des angiomes vertébraux. D’autres mutations du gène VHL puis des mutations dans les gènes PHD1, PHD2 et HIF2A ont ensuite été identifiées chez des patients atteints de polyglobulie congénitale. Les mutations du gène VHL responsables de polyglobulies sont moins délétères que celles décrites chez les patients avec maladie de VHL. Ces mutations entraînent en effet une faible activation de la voie de l’hypoxie, ce qui expliquerait l’absence de développement tumoral chez les patients polyglobuliques.12
Traitement des anémies
À l’inverse de la stratégie d’inhibition de la voie de l’hypoxie dans les cas de cancers, de polyglobulie ou d’HTAP, le traitement de certaines anémies liées à un défaut de production d’EPO nécessite l’activation de cette voie.
L’anémie est une complication majeure de la plupart des insuffisances rénales puisque le rein est la principale source de production d’EPO chez l’adulte. Le traitement par de l’EPO recombinante humaine a donc longtemps été un traitement de choix. Cependant, l’utilisation d’une forte quantité d’EPO pour stimuler suffisamment l’érythropoïèse des patients peut entraîner des effets indésirables : thrombose, accidents vasculaires cérébraux, insuffisance cardiaque, infarctus du myocarde et, dans de rares cas, production d’anticorps qui interagissent avec l’EPO endogène résiduelle et provoquent une aplasie érythrocytaire.
La stimulation de la production d’EPO endogène est donc une stratégie séduisante. De nombreux groupes de recherche ont étudié la réactivation de la production d’EPO par le foie, premier site de production lors de la vie fœtale. W. Kaelin a notamment mis en évidence que l’inactivation des PHD était suffisante pour réactiver la production d’EPO hépatique chez la souris, et les inhibiteurs des PHD ont prouvé leur efficacité dans le traitement des anémies liées à une insuffisance rénale chez l’homme. Trois inhibiteurs (daprodustat, roxadustat et vadadustat) sont maintenant en phase III de développement clinique et ont abouti à l’homologation récente du roxadustat pour le traitement de l’anémie en Chine.13 Ces inhibiteurs ont l’avantage de pouvoir être administrés oralement. Ils ont la capacité d’induire la production d’EPO endogène, gène cible de l’hypoxie qui semble le plus sensible à l’inhibition des PHD. Les effets secondaires des inhibiteurs de PHD sont encore mal connus car les études réalisées ont concerné un faible nombre de patients sur des périodes courtes, et la survenue d’éventuelles tumeurs est particulièrement surveillée,14 au vu du rôle de la voie de l’hypoxie dans l’oncogenèse.
L’anémie est une complication majeure de la plupart des insuffisances rénales puisque le rein est la principale source de production d’EPO chez l’adulte. Le traitement par de l’EPO recombinante humaine a donc longtemps été un traitement de choix. Cependant, l’utilisation d’une forte quantité d’EPO pour stimuler suffisamment l’érythropoïèse des patients peut entraîner des effets indésirables : thrombose, accidents vasculaires cérébraux, insuffisance cardiaque, infarctus du myocarde et, dans de rares cas, production d’anticorps qui interagissent avec l’EPO endogène résiduelle et provoquent une aplasie érythrocytaire.
La stimulation de la production d’EPO endogène est donc une stratégie séduisante. De nombreux groupes de recherche ont étudié la réactivation de la production d’EPO par le foie, premier site de production lors de la vie fœtale. W. Kaelin a notamment mis en évidence que l’inactivation des PHD était suffisante pour réactiver la production d’EPO hépatique chez la souris, et les inhibiteurs des PHD ont prouvé leur efficacité dans le traitement des anémies liées à une insuffisance rénale chez l’homme. Trois inhibiteurs (daprodustat, roxadustat et vadadustat) sont maintenant en phase III de développement clinique et ont abouti à l’homologation récente du roxadustat pour le traitement de l’anémie en Chine.13 Ces inhibiteurs ont l’avantage de pouvoir être administrés oralement. Ils ont la capacité d’induire la production d’EPO endogène, gène cible de l’hypoxie qui semble le plus sensible à l’inhibition des PHD. Les effets secondaires des inhibiteurs de PHD sont encore mal connus car les études réalisées ont concerné un faible nombre de patients sur des périodes courtes, et la survenue d’éventuelles tumeurs est particulièrement surveillée,14 au vu du rôle de la voie de l’hypoxie dans l’oncogenèse.
Références
1. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop- helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995;92:5510-4.
2. Richard S, Gardie B, Couvé S, Gad S. Von Hippel-Lindau: how a rare disease illuminates cancer biology. Semin Cancer Biol 2013;23:26-37.
3. Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822-6
4. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399:271-5.
5. Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001;292:468-72.
6. Ivan M, Kondo K, Yang H, et al. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292:464-8.
7. Epstein AC, Gleadle JM, McNeill LA,et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001;107:43-54.
8. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012:148:399-408.
9. Kaelin WG. New cancer targets emerging from studies of the Von Hippel-Lindau tumor suppressor protein. Ann N Y Acad Sci 2010;1210:1-7.
10. Cho H, Du X, Rizzi JP,et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016;539:107-11.
11. Chen W, Hill H, Christie A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016;539:112-7.
12. Lenglet M, Robriquet F, Schwarz K, et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018;132:469-83.
13. Dhillon S. Roxadustat: first global approval. Drugs 2019;79:563-72.
14. Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 2008;359:2685-92.
2. Richard S, Gardie B, Couvé S, Gad S. Von Hippel-Lindau: how a rare disease illuminates cancer biology. Semin Cancer Biol 2013;23:26-37.
3. Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822-6
4. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399:271-5.
5. Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001;292:468-72.
6. Ivan M, Kondo K, Yang H, et al. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292:464-8.
7. Epstein AC, Gleadle JM, McNeill LA,et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001;107:43-54.
8. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012:148:399-408.
9. Kaelin WG. New cancer targets emerging from studies of the Von Hippel-Lindau tumor suppressor protein. Ann N Y Acad Sci 2010;1210:1-7.
10. Cho H, Du X, Rizzi JP,et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016;539:107-11.
11. Chen W, Hill H, Christie A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016;539:112-7.
12. Lenglet M, Robriquet F, Schwarz K, et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018;132:469-83.
13. Dhillon S. Roxadustat: first global approval. Drugs 2019;79:563-72.
14. Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 2008;359:2685-92.