La responsabilité directe du changement de formulation dans la survenue de signes et symptômes indésirables chez certains patients est donc extrêmement probable.
À la demande des autorités, pour améliorer la stabilité du produit, une nouvelle formule de Levothyrox a été mise sur le marché en 2017 par le laboratoire Merck. De nombreux effets indésirables ont rapidement été signalés avec cette nouvelle formule. Le problème de la nouvelle formule de Levothyrox a été considéré, par le laboratoire Merck comme par les autorités sanitaires, essentiellement lié à un défaut d’information des patients. Une étude récente montre qu’il s’agit plutôt d’un défaut de formation du personnel du laboratoire et des autorités. En effet, en reprenant les données de l’étude de la bioéquivalence des deux formules,1 des statisticiens2 ont identifié une erreur dans l’analyse de ces données. Les données ont été mises en ligne,3, 4 ce qui permet de faire de cette histoire un cas d’école.
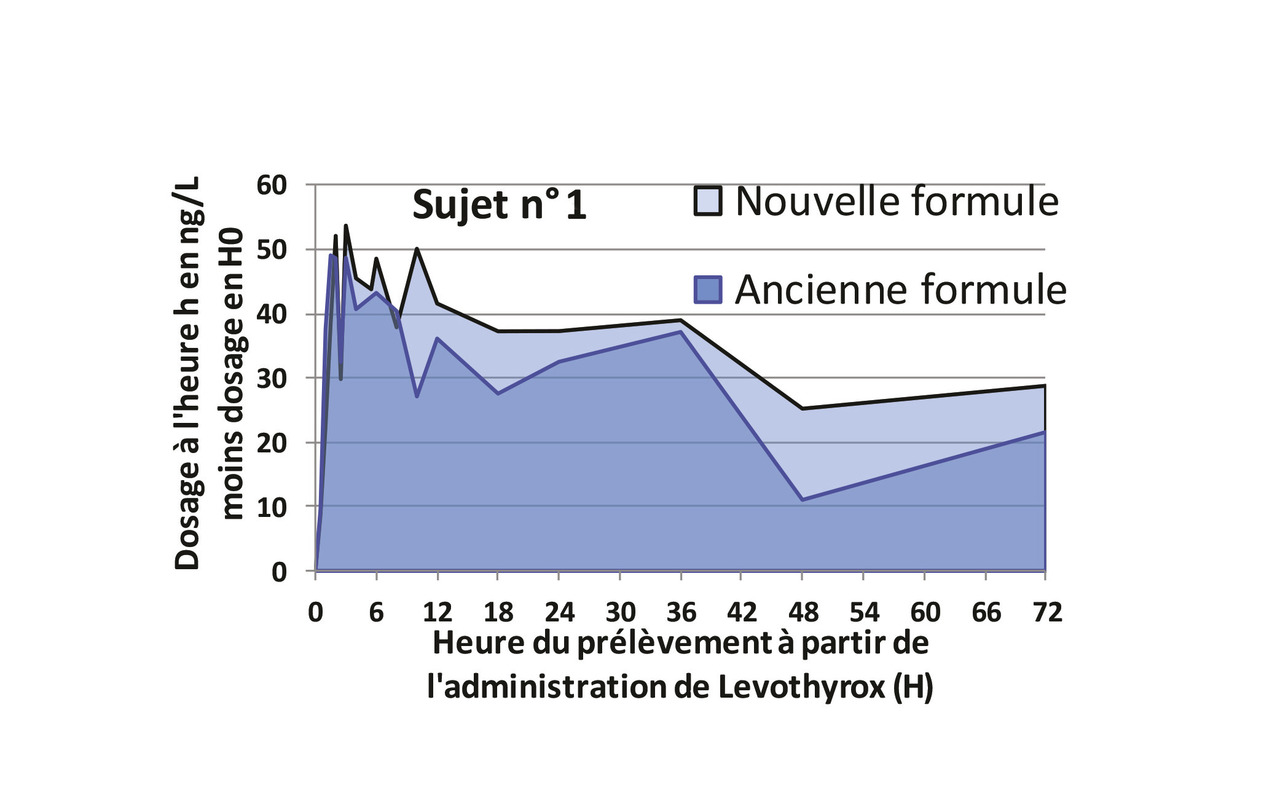
L’étude de la bioéquivalence a été faite en administrant à 204 volontaires sains les deux formules de Levothyrox, successivement, dans un ordre tiré au sort. Dix-sept prélèvements sanguins ont été réalisés, le premier avant l’administration du médicament (H0) et le dernier 72 heures plus tard (H72). Lafigure 1 montre, pour le premier sujet, les taux sériques de thyroxine aux 16 temps étudiés, après soustraction du taux au temps H0, reflet de la fonction thyroïdienne basale de chaque volontaire sain selon la recommandation des autorités*. À partir de ces données, on peut calculer, pour chacune des deux formulations, l’aire sous la courbe des taux de thyroxine après soustraction de la valeur en H0. Cette aire sous la courbe mesure directement la biodisponibilité de la lévothyroxine.
L’étude de la bioéquivalence a été faite en administrant à 204 volontaires sains les deux formules de Levothyrox, successivement, dans un ordre tiré au sort. Dix-sept prélèvements sanguins ont été réalisés, le premier avant l’administration du médicament (H0) et le dernier 72 heures plus tard (H72). La
L’analyse des biodisponibilités moyennes n’est pas concluante
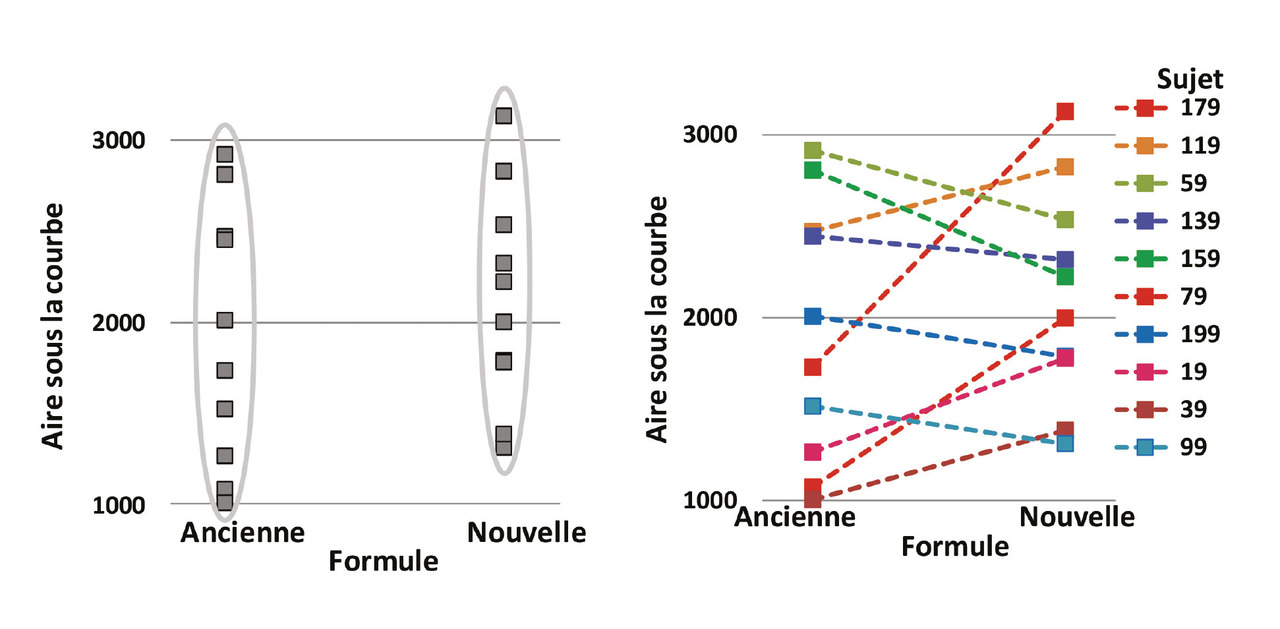
Le laboratoire Merck a soumis aux autorités une analyse comparant les biodisponibilités moyennes pour les 204 sujets avec la nouvelle et avec l’ancienne formulation1 et a conclu que la biodisponibilité de la nouvelle formule était équivalente à celle de l’ancienne formule. Cette analyse ne tient pas compte du fait que chaque sujet a reçu les deux formules, elle ne permet donc pas de vérifier que chacun de ces sujets répond de façon similaire aux deux formules ; on aurait fait la même analyse si on avait donné la nouvelle formule à 204 sujets et l’ancienne formule à 204 autres sujets comparables. L’analyse raisonnable consiste à étudier la différence des aires sous la courbe entre la nouvelle et l’ancienne formule chez un même sujet. Ainsi on compare la différence de biodisponibilités entre les deux formules pour chaque sujet. Pour conclure à la bioéquivalence entre les deux formules, les différences doivent être proches de zéro. La figure 1 montre, pour le sujet n° 1 de l’étude, l’évolution des dosages de thyroxine avec l’ancienne et la nouvelle formule. Le tableau et la figure 2 montrent les valeurs de l’aire sous la courbe pour dix patients. La figure 2A montre les données telles qu’elles ont été analysées par le laboratoire, qui s’est contenté de comparer les deux moyennes. La figure 2B montre les mêmes résultats présentés sujet par sujet.
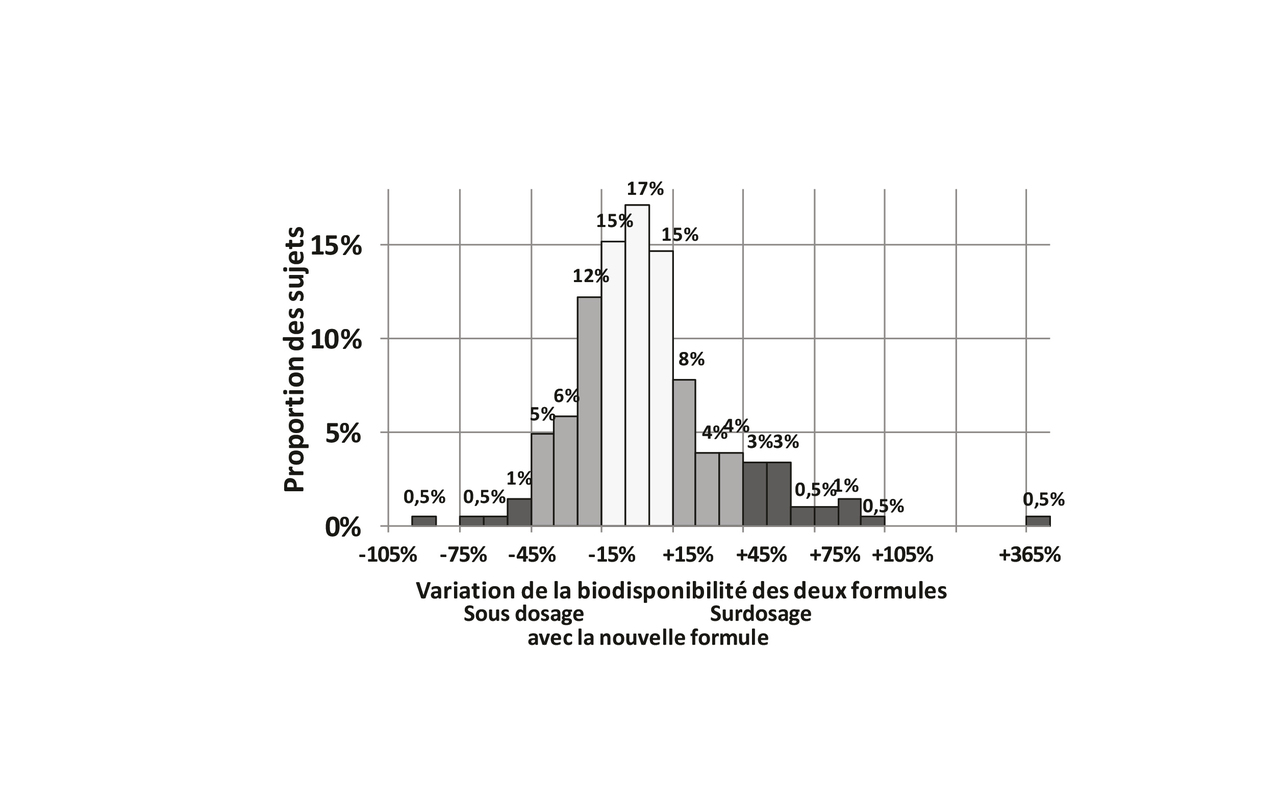
Pour les 204 sujets, les variations relatives de l’aire sous la courbe entre la nouvelle formule et l’ancienne formule vont de de -89 % à +100 % plus une valeur extrême de 369 %, montrant une variabilité importante de la biodisponibilité entre les deux formules d’un sujet à l’autre. Lafigure 3 montre que 26 % des sujets ont un sous-dosage d’au moins 15 % avec la nouvelle formule, que 47 % n’ont pas plus de 15 % d’écart, et que 27 % ont un surdosage d’au moins 15 % avec la nouvelle formule. Le sous-dosage est de plus de 45 % pour 2,9 % des sujets et le surdosage est d’au moins 45 % pour 11 % des sujets. Et c’est ainsi que l’on met en évidence une variabilité importante de la biodisponibilité de la nouvelle formule d’un sujet à l’autre. L’usage étant d’ajuster la posologie journalière, qui est généralement comprise entre 50 et 150 μg, en procédant par paliers de 25 μg/j ou même de 12,5 μg/j, un surdosage soudain de 45 % est considérable.
Naturellement, cette étude sur des sujets sains ne démontre pas l’existence d’un lien entre sur- ou sous-dosage et symptômes indésirables chez les patients. La comparaison des biodisponibilités individuelles chez chaque sujet montre seulement que le changement de formule a entraîné une différence de biodisponibilité importante chez certains sujets. La responsabilité directe du changement de formulation dans la survenue de signes et symptômes indésirables chez certains patients est donc extrêmement probable.
Au lieu de dire aux patients qui se sont plaints que leurs symptômes indésirables avaient pour origine un défaut d’information, on aurait dû les prévenir de la possible différence de biodisponibilité des deux formules du médicament et leur recommander de consulter rapidement s’ils ne se sentaient pas bien.
Pour les 204 sujets, les variations relatives de l’aire sous la courbe entre la nouvelle formule et l’ancienne formule vont de de -89 % à +100 % plus une valeur extrême de 369 %, montrant une variabilité importante de la biodisponibilité entre les deux formules d’un sujet à l’autre. La
Naturellement, cette étude sur des sujets sains ne démontre pas l’existence d’un lien entre sur- ou sous-dosage et symptômes indésirables chez les patients. La comparaison des biodisponibilités individuelles chez chaque sujet montre seulement que le changement de formule a entraîné une différence de biodisponibilité importante chez certains sujets. La responsabilité directe du changement de formulation dans la survenue de signes et symptômes indésirables chez certains patients est donc extrêmement probable.
Au lieu de dire aux patients qui se sont plaints que leurs symptômes indésirables avaient pour origine un défaut d’information, on aurait dû les prévenir de la possible différence de biodisponibilité des deux formules du médicament et leur recommander de consulter rapidement s’ils ne se sentaient pas bien.
Des autorités peu exigeantes
La première auteure de l’analyse réalisée pour le compte de Merck, interrogée par mail il y a plus d’un an, avait répondu que l’analyse réalisée était celle qui était demandée par les autorités, et cela est confirmé par l’équipe de D. Concordet.2 Ainsi, il semble que les autorités aient encouragé les statisticiens de Merck à faire une analyse suboptimale en comparant les biodisponibilités moyennes observées avec chaque formule, ce qui ne met pas en évidence de différence de biodisponibilité.
On peut aussi rapprocher cette histoire de diverses autres erreurs faites par les laboratoires pharmaceutiques, erreurs dont ils se sont défendus en s’appuyant sur les contraintes réglementaires.
C’est ainsi qu’après l’essai de phase I du TGN1412 en 2006 qui a conduit six jeunes volontaires sains en réanimation à Londres, et à l’amputation des doigts de main et de pied chez l’un d’entre eux, on a continué à administrer simultanément à plusieurs personnes une nouvelle molécule à une dose jamais étudiée chez l’homme parce que la règlementation est restée vague, donc sujette à interprétation.5 En effet, l’agence européenne a seulement requis un examen des données précliniques pour décider si la première dose qu’il est prévu d’administrer à un humain est raisonnable et des règles d’arrêt si les choses ne se déroulent pas comme prévu**. Ainsi, si les données précliniques ne sont pas inquiétantes, il n’est pas prévu d’étaler l’inclusion des sujets dans le temps. Après le décès d’un volontaire sain et les séquelles d’autres volontaires sains traités simultanément en 2015 chez Biotrial à Rennes, la règle demeure inchangée.6
C’est ainsi également que Sanofi déclare avoir suivi à la lettre les recommandations des autorités sur la prescription de valproate de sodium (Dépakine, Dépamide, Dépakote) aux femmes en âge de procréer, alors que de nombreux signaux indiquaient que cette prescription conduit dans 10 % des cas à la naissance d’un enfant ayant une malformation, et dans 30 % à 40 % des cas à la naissance d’un enfant ayant des troubles neurodévelopementaux.7
Et donc Merck persiste dans cette stratégie en expliquant que l’équivalence des deux formulations de Levothyrox a été étudiée selon les demandes des autorités.
On peut aussi rapprocher cette histoire de diverses autres erreurs faites par les laboratoires pharmaceutiques, erreurs dont ils se sont défendus en s’appuyant sur les contraintes réglementaires.
C’est ainsi qu’après l’essai de phase I du TGN1412 en 2006 qui a conduit six jeunes volontaires sains en réanimation à Londres, et à l’amputation des doigts de main et de pied chez l’un d’entre eux, on a continué à administrer simultanément à plusieurs personnes une nouvelle molécule à une dose jamais étudiée chez l’homme parce que la règlementation est restée vague, donc sujette à interprétation.5 En effet, l’agence européenne a seulement requis un examen des données précliniques pour décider si la première dose qu’il est prévu d’administrer à un humain est raisonnable et des règles d’arrêt si les choses ne se déroulent pas comme prévu**. Ainsi, si les données précliniques ne sont pas inquiétantes, il n’est pas prévu d’étaler l’inclusion des sujets dans le temps. Après le décès d’un volontaire sain et les séquelles d’autres volontaires sains traités simultanément en 2015 chez Biotrial à Rennes, la règle demeure inchangée.6
C’est ainsi également que Sanofi déclare avoir suivi à la lettre les recommandations des autorités sur la prescription de valproate de sodium (Dépakine, Dépamide, Dépakote) aux femmes en âge de procréer, alors que de nombreux signaux indiquaient que cette prescription conduit dans 10 % des cas à la naissance d’un enfant ayant une malformation, et dans 30 % à 40 % des cas à la naissance d’un enfant ayant des troubles neurodévelopementaux.7
Et donc Merck persiste dans cette stratégie en expliquant que l’équivalence des deux formulations de Levothyrox a été étudiée selon les demandes des autorités.
* Certains médecins contestent cette soustraction. Selon l’Agence européenne du médicament, « si la substance étudiée est endogène, le calcul des paramètres pharmacocinétiques doit être fait en corrigeant pour la valeur basale de façon à ce que ces paramètres reflètent les concentrations supplémentaires dues au traitement » (“If the substance being studied is endogenous, the calculation of pharmacokinetic parameters should be performed using baseline correction so that the calculated pharmacokinetic parameters refer to the additional concentrations provided by the treatment”). https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf ou http://bit.ly/2EPEHfF ** The MHRA [Medicines and Health products Regulatory Agency] now ensures committees look at pre-clinical data, to decide whether the first dose given to humans is the right dose and has rules for stopping if things don't go as expected.Voir aussi l'édito p. 583.
1. Gottwald-Hostalek U, Uhl W, Wolna P, Kahaly GJ. New levothyroxine formulation meeting 95-105% specification over the whole shelf-life: results from two pharmacokinetic trials. Curr Med Res Opin. 2017;33:169-74.
2. Concordet D, Gandia P, Montastruc JL, et al. Levothyrox® new and old formulations: are they switchable for millions of patients? Clin Pharmacokinet. 2019 Apr 4. doi: 10.1007/s40262-019-00747-3.
3. Agence nationale du médicament et des produits de santé. Levothyrox. http://bit.ly/2WpwycZ
4. Agence nationale du médicament et des produits de santé. Levothyrox. Bioequivalence trial of new Levothyrox formulation versus old formulation. http://bit.ly/2IkcM8R (page 442)
5. Hill C, Asselain B. More on the phase 1 trial of BIA 10-2474, conducted by Biotrial in Rennes and during which a volunteer died. Letter on the article "The BIAL/Biotrial case of death of a human volunteer in the first-in-human study of BIA 10-2474: Are we missing the fundamentals?''. Presse Med 2017;46:349-52. http://bit.ly/2WLjHB6
6. European medicines agency. Revised guideline on first-in-human clinical trials. EMA, 2017. http://bit.ly/2wCdubQ
7. Agence nationale du médicament et des produits de santé. Valproate et dérivés : contre-indication pendant la grossesse (sauf situations exceptionnelles) et programme de prévention des grossesses. Lettre aux professionnels de santé, ANSM 2018. http://bit.ly/2EPGNw3
2. Concordet D, Gandia P, Montastruc JL, et al. Levothyrox® new and old formulations: are they switchable for millions of patients? Clin Pharmacokinet. 2019 Apr 4. doi: 10.1007/s40262-019-00747-3.
3. Agence nationale du médicament et des produits de santé. Levothyrox. http://bit.ly/2WpwycZ
4. Agence nationale du médicament et des produits de santé. Levothyrox. Bioequivalence trial of new Levothyrox formulation versus old formulation. http://bit.ly/2IkcM8R (page 442)
5. Hill C, Asselain B. More on the phase 1 trial of BIA 10-2474, conducted by Biotrial in Rennes and during which a volunteer died. Letter on the article "The BIAL/Biotrial case of death of a human volunteer in the first-in-human study of BIA 10-2474: Are we missing the fundamentals?''. Presse Med 2017;46:349-52. http://bit.ly/2WLjHB6
6. European medicines agency. Revised guideline on first-in-human clinical trials. EMA, 2017. http://bit.ly/2wCdubQ
7. Agence nationale du médicament et des produits de santé. Valproate et dérivés : contre-indication pendant la grossesse (sauf situations exceptionnelles) et programme de prévention des grossesses. Lettre aux professionnels de santé, ANSM 2018. http://bit.ly/2EPGNw3