Quelques repères diagnostiques cliniques et paracliniques utilisables en pratique quotidienne permettent de ne pas méconnaître une encéphalite auto-immune.
Les encéphalites auto-immunes sont des maladies rares du système nerveux central qui atteignent de façon prédominante les structures limbiques (hippocampe, amygdale) et sont associées à des auto-anticorps ciblant des protéines neuronales ou gliales.
Les protéines ciblées se situent le plus souvent à la surface membranaire des neurones, par exemple par les anticorps anti-NMDAR, antiLGI1, anti-CASPR2, ou IgLON5. Ces anticorps perturbent directement les fonctions neuronales ; les symptômes sont donc souvent réversibles sous immunosuppresseurs.
Certaines encéphalites auto-immunes sont, au contraire, associées à des anticorps qui ciblent des protéines intracellulaires ; elles sont souvent paranéoplasiques, et leur pronostic est péjoratif.1
Les protéines ciblées se situent le plus souvent à la surface membranaire des neurones, par exemple par les anticorps anti-NMDAR, antiLGI1, anti-CASPR2, ou IgLON5. Ces anticorps perturbent directement les fonctions neuronales ; les symptômes sont donc souvent réversibles sous immunosuppresseurs.
Certaines encéphalites auto-immunes sont, au contraire, associées à des anticorps qui ciblent des protéines intracellulaires ; elles sont souvent paranéoplasiques, et leur pronostic est péjoratif.1
Une présentation clinique qui traduit l’atteinte limbique
Les encéphalites auto-immunes s’installent généralement de façon subaiguë (en moins de trois mois), parfois encore plus progressivement (tableau 1 ).2 Les manifestations cliniques les plus fréquentes traduisent l’atteinte du système limbique, impliqué dans la mémoire et les émotions : amnésie antérograde, troubles comportementaux, et épilepsie focale temporale interne.2
L’amnésie antérograde se caractérise par un oubli à mesure, avec une relative préservation des souvenirs anciens. Une amnésie rétrograde, couvrant une période de plusieurs années, peut lui être associée. Les troubles comportementaux sont liés à un dysfonctionnement des réseaux frontaux, fortement associés au système limbique, avec irritabilité, impulsivité, désinhibition, ou au contraire apathie et ralentissement psychomoteur. Les crises temporales internes se présentent typiquement par une rupture de contact, des automatismes moteurs, ou une aura épigastrique ascendante, parfois des troubles de mémoire paroxystiques ou des troubles du langage. Elles peuvent être généralisées d’emblée. En plus de ces manifestations limbiques, des signes extralimbiques peuvent être présents, comme des mouvements anormaux (chorée, dyskinésies), un syndrome cérébelleux, ou des signes neurovégétatifs (tableau 1 ).3
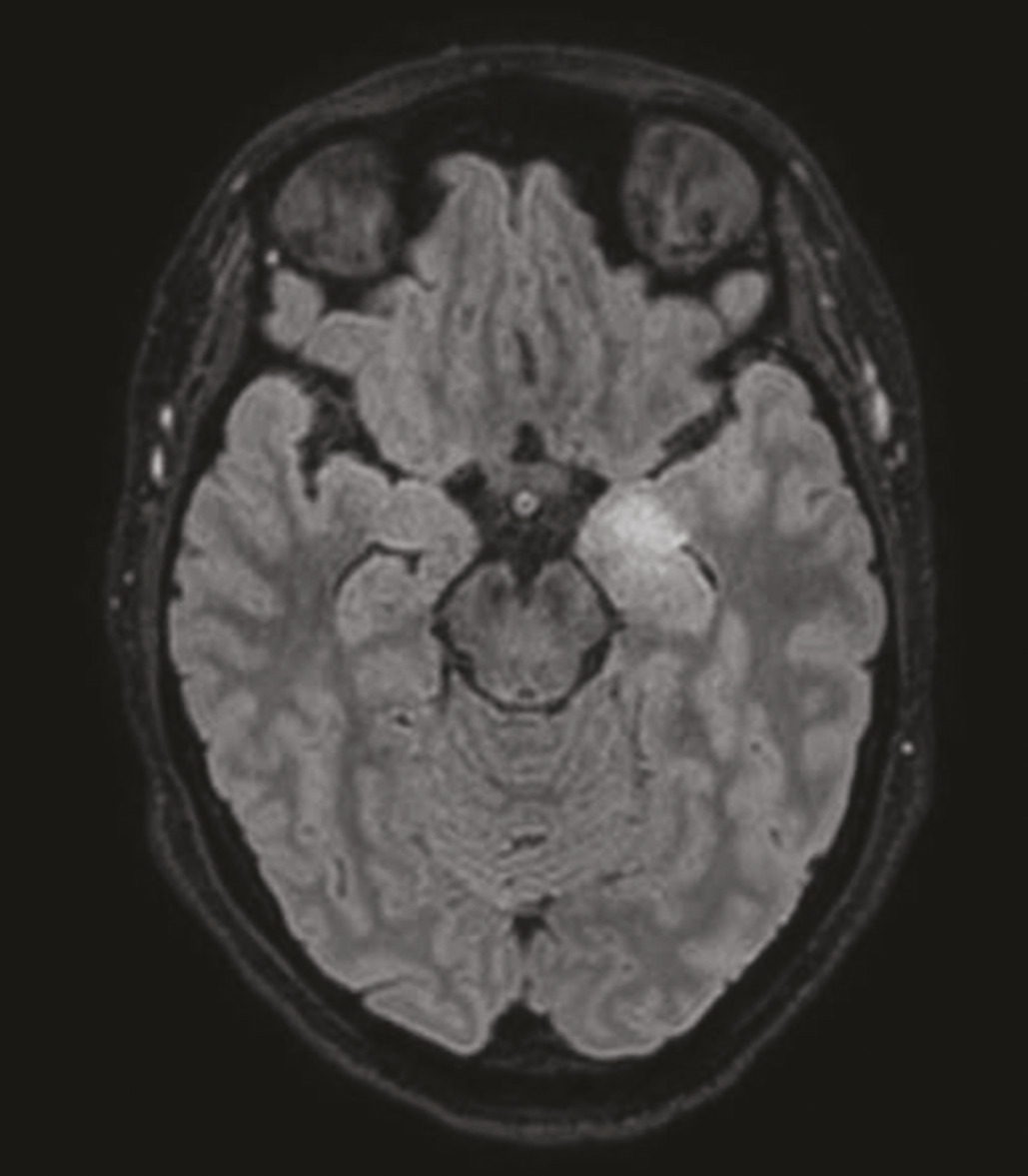
L’imagerie par résonance magnétique (IRM) cérébrale peut montrer un hypersignal T2 temporal interne unilatéral ou bilatéral ne prenant pas le contraste (fig. 1 ), mais peut également être normale.2, 3 L’électroencéphalogramme (EEG) peut mettre en évidence une focalisation aux régions temporales (ralentissement, activité paroxystique).2 L’étude du liquide cérébrospinal (LCS) peut trouver des signes d’inflammation (pléiocytose, bandes oligoclonales) ou être normale.3
L’amnésie antérograde se caractérise par un oubli à mesure, avec une relative préservation des souvenirs anciens. Une amnésie rétrograde, couvrant une période de plusieurs années, peut lui être associée. Les troubles comportementaux sont liés à un dysfonctionnement des réseaux frontaux, fortement associés au système limbique, avec irritabilité, impulsivité, désinhibition, ou au contraire apathie et ralentissement psychomoteur. Les crises temporales internes se présentent typiquement par une rupture de contact, des automatismes moteurs, ou une aura épigastrique ascendante, parfois des troubles de mémoire paroxystiques ou des troubles du langage. Elles peuvent être généralisées d’emblée. En plus de ces manifestations limbiques, des signes extralimbiques peuvent être présents, comme des mouvements anormaux (chorée, dyskinésies), un syndrome cérébelleux, ou des signes neurovégétatifs (
L’imagerie par résonance magnétique (IRM) cérébrale peut montrer un hypersignal T2 temporal interne unilatéral ou bilatéral ne prenant pas le contraste (
L’encéphalite à anticorps anti-NMDAR très stéréotypée
Avec 50 à 60 cas par an, en France, il s’agit de l’encéphalite auto-immune la plus fréquente.4 Elle atteint dans 80 % des cas la femme, à un âge médian de 21 ans. La population pédiatrique comptabilise 40 % des malades, et seuls 5 % des patients ont plus de 45 ans.5 Un tératome ovarien est présent dans environ 40 % des cas.1
La présentation clinique est à connaître, car très stéréotypée, avec de nombreux signes non limbiques associés. Les prodromes infectieux sont fréquents, et précèdent de quelques semaines l’installation de bruyants symptômes psychiatriques.6 Il s’agit le plus souvent d’un état délirant non systématisé, aux thématiques multiples et polymorphes, souvent mystiques, mégalomaniaques et de persécution. Les hallucinations visuelles et acoustico-verbales sont fréquentes. Des troubles de l’humeur de type anxiété, irritabilité, symptômes maniaques ou dépressifs s’associent dans près de la moitié des cas. Une insomnie est présente chez au moins 20 % des patients.6
La majorité des patients sont ainsi d’abord admis en psychiatrie, avant que les signes neurologiques n’apparaissent, classiquement dans les semaines qui suivent. Il s’agit alors de crises épileptiques généralisées ou focales, ou de mouvements anormaux, comme des dyskinésies buccofaciales ou de la distalité des membres, parfois une dystonie.3 Les troubles psychiatriques peuvent alors laisser la place à des signes plus franchement cognitifs, comme l’amnésie antérograde et le syndrome frontal dysexécutif.
Enfin, dans plus de deux tiers des cas apparaissent des troubles graves du système nerveux autonome tels que des anomalies rythmiques et tensionnelles, une hyperthermie, et/ou une hypoventilation centrale, justifiant une prise en charge en réanimation.5
Les états de mal épileptique sont aussi possibles à ce stade. Le début est différent chez l’homme et dans la population pédiatrique, où l’épilepsie est le mode d’entrée classique, avec peu de symptômes psychotiques ; à la phase d’état, en revanche, les présentations sont similaires quels que soient l’âge et le sexe.2
L’IRM cérébrale visualise un hypersignal temporal interne dans seulement un tiers des cas (fig. 1 ). L’EEG peut révéler des signes évocateurs, comme l’extreme delta brush*, notamment dans les formes sévères, mais le plus souvent le tracé est simplement ralenti et désorganisé.2
La ponction lombaire montre une lymphocytose dans deux tiers des cas. Le diagnostic est retenu sur positivité des anticorps anti-NMDAR dans le LCS, la recherche dans le sérum étant moins sensible et spécifique.2, 3
La mortalité est de 5 à 10 % selon les séries, en lien avec les complications de la phase d’état (état de mal, hypoventilation centrale). Sous traitement, l’évolution est habituellement lentement favorable, en dehors de rares formes réfractaires. L’amélioration est retardée, débutant en moyenne après deux à trois mois de traitement, et très progressive. Malgré la fréquente sévérité initiale de cette maladie, la récupération complète, ou avec de minimes séquelles (troubles de la concentration essentiellement), est de 80 % à deux ans.5
La présentation clinique est à connaître, car très stéréotypée, avec de nombreux signes non limbiques associés. Les prodromes infectieux sont fréquents, et précèdent de quelques semaines l’installation de bruyants symptômes psychiatriques.6 Il s’agit le plus souvent d’un état délirant non systématisé, aux thématiques multiples et polymorphes, souvent mystiques, mégalomaniaques et de persécution. Les hallucinations visuelles et acoustico-verbales sont fréquentes. Des troubles de l’humeur de type anxiété, irritabilité, symptômes maniaques ou dépressifs s’associent dans près de la moitié des cas. Une insomnie est présente chez au moins 20 % des patients.6
La majorité des patients sont ainsi d’abord admis en psychiatrie, avant que les signes neurologiques n’apparaissent, classiquement dans les semaines qui suivent. Il s’agit alors de crises épileptiques généralisées ou focales, ou de mouvements anormaux, comme des dyskinésies buccofaciales ou de la distalité des membres, parfois une dystonie.3 Les troubles psychiatriques peuvent alors laisser la place à des signes plus franchement cognitifs, comme l’amnésie antérograde et le syndrome frontal dysexécutif.
Enfin, dans plus de deux tiers des cas apparaissent des troubles graves du système nerveux autonome tels que des anomalies rythmiques et tensionnelles, une hyperthermie, et/ou une hypoventilation centrale, justifiant une prise en charge en réanimation.5
Les états de mal épileptique sont aussi possibles à ce stade. Le début est différent chez l’homme et dans la population pédiatrique, où l’épilepsie est le mode d’entrée classique, avec peu de symptômes psychotiques ; à la phase d’état, en revanche, les présentations sont similaires quels que soient l’âge et le sexe.2
L’IRM cérébrale visualise un hypersignal temporal interne dans seulement un tiers des cas (
La ponction lombaire montre une lymphocytose dans deux tiers des cas. Le diagnostic est retenu sur positivité des anticorps anti-NMDAR dans le LCS, la recherche dans le sérum étant moins sensible et spécifique.2, 3
La mortalité est de 5 à 10 % selon les séries, en lien avec les complications de la phase d’état (état de mal, hypoventilation centrale). Sous traitement, l’évolution est habituellement lentement favorable, en dehors de rares formes réfractaires. L’amélioration est retardée, débutant en moyenne après deux à trois mois de traitement, et très progressive. Malgré la fréquente sévérité initiale de cette maladie, la récupération complète, ou avec de minimes séquelles (troubles de la concentration essentiellement), est de 80 % à deux ans.5
Les crises dystoniques brachiofaciales pathognomoniques de l’encéphalite à anti-LGI1
L’encéphalite à anticorps anti-LGI1 est la deuxième encéphalite auto-immune la plus fréquente en France (50 cas par an).4 L’âge moyen est de 60 à 65 ans, avec une prédominance masculine (50-65 %).7
Les symptômes s’installent habituellement en plusieurs semaines, mais l’évolution peut être progressive sur plusieurs mois. Les troubles cognitifs et comportementaux sont fréquents et sévères, dans la majorité des cas des troubles mnésiques antérogrades et une atteinte dysexécutive. Des éléments dépressifs ou anxieux peuvent exister.7
Les crises épileptiques focales temporales internes sont fréquentes. On peut aussi observer des crises dystoniques brachiofaciales, qui sont pathognomoniques de cette entité.8 Elles se caractérisent par une contraction brève du membre supérieur, parfois de l’hémiface homolatérale, voire du membre inférieur. Elles sont extrêmement fréquentes (plusieurs dizaines ou centaines d’épisodes par jour), peuvent être unilatérales ou bilatérales à bascule, et persistent pendant le sommeil. Enfin, il peut exister des troubles du comportement en sommeil paradoxal et une insomnie.
Sur le plan biologique, l’hyponatrémie par sécrétion inappropriée d’hormone antidiurétique est fréquente et évocatrice.2, 8 Le diagnostic repose sur la mise en évidence des anticorps anti-LGI1, qui doivent être recherchés dans le LCS et le sérum.2,3,8 La réponse de l’épilepsie et des crises dystoniques au traitement immunosuppresseur, en particulier aux corticoïdes, est souvent bonne. En revanche, la majorité des patients conservent des séquelles cognitives légères à modérées.7 Le pronostic est intimement lié à la précocité du diagnostic et à la mise en place de thérapeutiques adaptées.3, 7, 8
Les symptômes s’installent habituellement en plusieurs semaines, mais l’évolution peut être progressive sur plusieurs mois. Les troubles cognitifs et comportementaux sont fréquents et sévères, dans la majorité des cas des troubles mnésiques antérogrades et une atteinte dysexécutive. Des éléments dépressifs ou anxieux peuvent exister.7
Les crises épileptiques focales temporales internes sont fréquentes. On peut aussi observer des crises dystoniques brachiofaciales, qui sont pathognomoniques de cette entité.8 Elles se caractérisent par une contraction brève du membre supérieur, parfois de l’hémiface homolatérale, voire du membre inférieur. Elles sont extrêmement fréquentes (plusieurs dizaines ou centaines d’épisodes par jour), peuvent être unilatérales ou bilatérales à bascule, et persistent pendant le sommeil. Enfin, il peut exister des troubles du comportement en sommeil paradoxal et une insomnie.
Sur le plan biologique, l’hyponatrémie par sécrétion inappropriée d’hormone antidiurétique est fréquente et évocatrice.2, 8 Le diagnostic repose sur la mise en évidence des anticorps anti-LGI1, qui doivent être recherchés dans le LCS et le sérum.2,3,8 La réponse de l’épilepsie et des crises dystoniques au traitement immunosuppresseur, en particulier aux corticoïdes, est souvent bonne. En revanche, la majorité des patients conservent des séquelles cognitives légères à modérées.7 Le pronostic est intimement lié à la précocité du diagnostic et à la mise en place de thérapeutiques adaptées.3, 7, 8
L’encéphalite à anti-CASPR2 répond bien au traitement
L’encéphalite auto-immune à anti-CASPR2 se caractérise par un mode d’installation très progressif et une grande variété de symptômes dont les plus fréquents sont l’épilepsie temporale, l’ataxie cérébelleuse, parfois paroxystique, les troubles mnésiques et dysexécutifs, l’altération de l’état général, les troubles de l’humeur et du sommeil, et les douleurs neurogènes.9 Ces symptômes s’installent de façon séquentielle et ne sont pas tous présents au début.
Le LCS est anormal dans plus de 60 % des cas, avec une pléiocytose et/ou des bandes oligoclonales, de même que l’IRM cérébrale, avec des hypersignaux temporaux internes. Du fait de sa présentation polymorphe et lentement progressive, l’encéphalite anti-CASPR2 semble largement sous-diagnostiquée. L’identification de ces anticorps est pourtant fondamentale, car la majorité des patients récupèrent sous traitement.
Le LCS est anormal dans plus de 60 % des cas, avec une pléiocytose et/ou des bandes oligoclonales, de même que l’IRM cérébrale, avec des hypersignaux temporaux internes. Du fait de sa présentation polymorphe et lentement progressive, l’encéphalite anti-CASPR2 semble largement sous-diagnostiquée. L’identification de ces anticorps est pourtant fondamentale, car la majorité des patients récupèrent sous traitement.
L’épilepsie temporale interne au premier plan de l’encéphalite à anti-GAD65
Les anticorps anti-GAD65, bien qu’initialement décrits dans le diabète auto-immun, sont également associés à trois syndromes neurologiques auto-immuns : la cérébellite auto-immune, le syndrome de la personne raide et l’encéphalite auto-immune.10 Dans ces syndromes neurologiques, les anticorps anti-GAD65 sont dosés à titres très élevés et sont détectables dans le LCS, à la différence du diabète.
L’encéphalite auto-immune à anticorps anti-GAD65 peut se déclarer à tout âge, avec une prédominance féminine. Il existe souvent un terrain auto-immun personnel ou familial.3 L’installation des symptômes peut être subaiguë ou, au contraire, très insidieuse. L’épilepsie temporale interne est au premier plan, souvent pharmacorésistante.10 Elle s’accompagne de troubles mnésiques et dysexécutifs sévères, qui peuvent être aggravés par des crises épileptiques infracliniques. L’IRM est normale dans plus de 50 % des cas. L’étude du LCS révèle presque constamment des bandes oligoclonales surnuméraires.2,3,10 Le pronostic est péjoratif, sans doute aggravé par le fréquent retard diagnostique.10
L’encéphalite auto-immune à anticorps anti-GAD65 peut se déclarer à tout âge, avec une prédominance féminine. Il existe souvent un terrain auto-immun personnel ou familial.3 L’installation des symptômes peut être subaiguë ou, au contraire, très insidieuse. L’épilepsie temporale interne est au premier plan, souvent pharmacorésistante.10 Elle s’accompagne de troubles mnésiques et dysexécutifs sévères, qui peuvent être aggravés par des crises épileptiques infracliniques. L’IRM est normale dans plus de 50 % des cas. L’étude du LCS révèle presque constamment des bandes oligoclonales surnuméraires.2,3,10 Le pronostic est péjoratif, sans doute aggravé par le fréquent retard diagnostique.10
Encéphalite à anti-IgLON5 : un pronostic sombre
Les encéphalites associées aux anticorps anti-IgLON5 sont plus rares mais probablement sous-diagnostiquées.2,3 Leur installation est très progressive, sur un mode pseudodégénératif. La symptomatologie est hétérogène, avec l’association variable d’un syndrome cérébelleux, de troubles cognitifs, de troubles du sommeil, de mouvements anormaux choréiques et/ou d’une atteinte bulbaire avec stridor nocturne.
L’IRM cérébrale et l’analyse du LCS sont habituellement normales, et le diagnostic repose sur la détection d’anticorps anti-IgLON5 dans le LCS.2 Le pronostic est sombre, la majorité des patients décèdent ou gardent de lourdes séquelles.3 Quelques encéphalites à anticorps anti-IgLON5 répondent toutefois aux immunosuppresseurs.
L’IRM cérébrale et l’analyse du LCS sont habituellement normales, et le diagnostic repose sur la détection d’anticorps anti-IgLON5 dans le LCS.2 Le pronostic est sombre, la majorité des patients décèdent ou gardent de lourdes séquelles.3 Quelques encéphalites à anticorps anti-IgLON5 répondent toutefois aux immunosuppresseurs.
La plupart des encéphalites paranéoplasiques précède la découverte du cancer
Les encéphalites auto-immunes paranéoplasiques sont essentiellement associées à des anticorps ciblant des protéines intracellulaires (tableau 2 ).1 Il s’agit d’encéphalites limbiques (amnésie antérograde, troubles comportementaux, épilepsie temporale), auxquelles peuvent s’associer, selon l’anticorps, d’autres syndromes neurologiques paranéoplasiques (neuronopathie sensitive subaiguë, pseudo-obstruction intestinale chronique, ou encore ataxie cérébelleuse).
L’IRM cérébrale, l’étude du LCS et l’EEG peuvent être normaux, ce qui ne doit pas écarter le diagnostic.2
L’évolution est aiguë ou subaiguë (moins de trois mois) et monophasique. Le tableau neurologique précède la découverte du cancer dans la majorité des cas. Lorsqu’un cancer est déjà connu, l’encéphalite paranéoplasique peut signaler une récidive tumorale. Par ailleurs, les immunothérapies anticancéreuses par inhibiteurs de checkpoints semblent favoriser, dans de rares cas, l’émergence d’encéphalites auto-immunes paranéoplasiques.1-3
Le diagnostic repose sur la mise en évidence d’un anticorps antineuronal sérique et/ou du LCS, associé à un tableau clinique et un cancer compatibles.1 L’évolution est généralement péjorative, avec peu ou pas de récupération après la stabilisation clinique. La précocité du traitement du cancer et des traitements immunomodulateurs semble fondamentale pour stabiliser la dégradation neurologique.
L’IRM cérébrale, l’étude du LCS et l’EEG peuvent être normaux, ce qui ne doit pas écarter le diagnostic.2
L’évolution est aiguë ou subaiguë (moins de trois mois) et monophasique. Le tableau neurologique précède la découverte du cancer dans la majorité des cas. Lorsqu’un cancer est déjà connu, l’encéphalite paranéoplasique peut signaler une récidive tumorale. Par ailleurs, les immunothérapies anticancéreuses par inhibiteurs de checkpoints semblent favoriser, dans de rares cas, l’émergence d’encéphalites auto-immunes paranéoplasiques.1-3
Le diagnostic repose sur la mise en évidence d’un anticorps antineuronal sérique et/ou du LCS, associé à un tableau clinique et un cancer compatibles.1 L’évolution est généralement péjorative, avec peu ou pas de récupération après la stabilisation clinique. La précocité du traitement du cancer et des traitements immunomodulateurs semble fondamentale pour stabiliser la dégradation neurologique.
Une démarche diagnostique bien établie
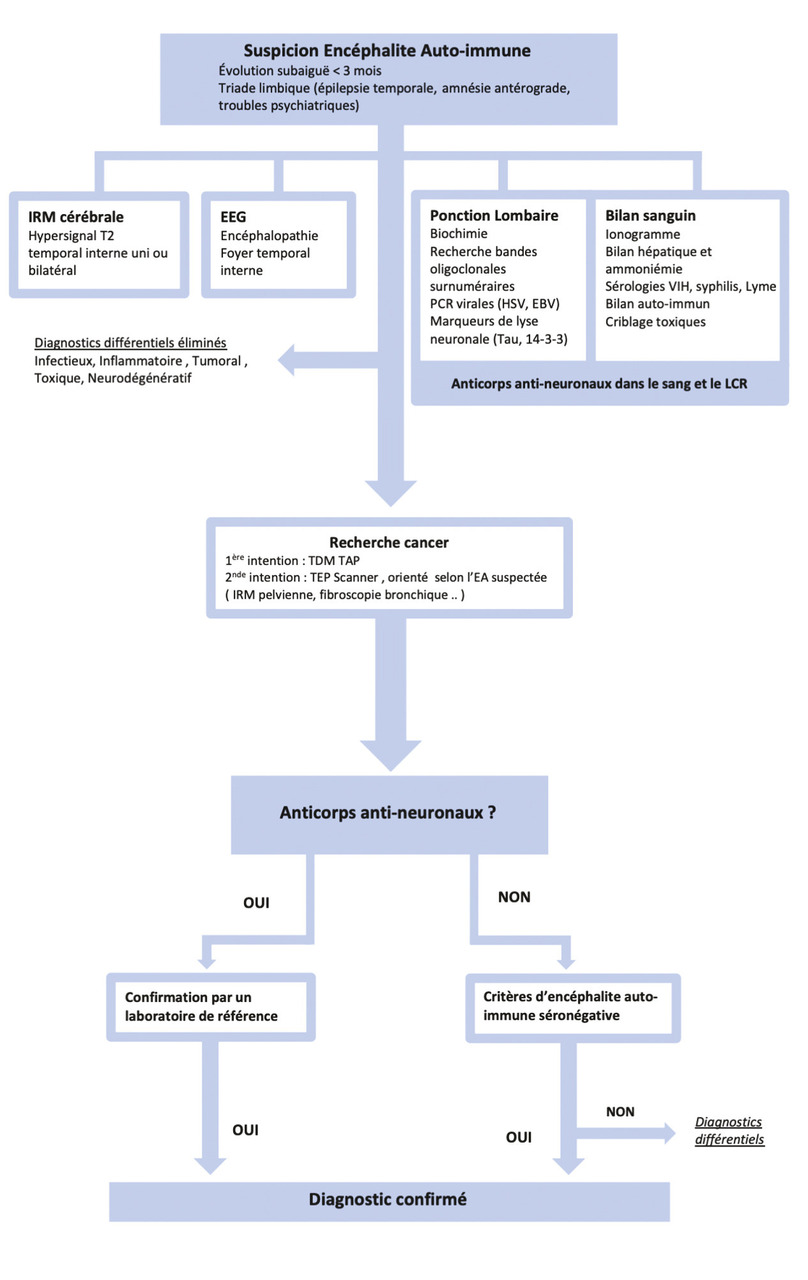
L’apparition aiguë ou subaiguë, en moins de trois mois, de symptômes de la triade limbique doit faire évoquer le diagnostic d’encéphalite auto-immune (fig. 2 ).2 L’évolution peut toutefois être plus progressive. Un bilan minimal doit être réalisé, à la recherche des principaux diagnostics différentiels que sont les pathologies tumorales, infectieuses, inflammatoires, toxiques et neurodégénératives.2
Le bilan comprend une IRM cérébrale, un EEG et une ponction lombaire. Si l’IRM révèle des hypersignaux hippocampiques, le principal diagnostic différentiel est le gliome, et le moindre doute sur un aspect infiltrant ou une prise de contraste impose une surveillance radioclinique rapprochée. Les examens biologiques doivent comporter une recherche £de troubles métaboliques et, selon le contexte, un bilan sérologique infectieux et un criblage des toxiques.2 La réalisation de PCR virales dans le liquide cérébrospinal (herpès simplex virus, virus d’Epstein-Barr, virus varicelle-zona) est primordiale. Enfin, l’évaluation des marqueurs de neurodégénérescence (Tau, phospho-Tau, protéine bêta-amyloïde) peut aider à distinguer une encéphalite auto-immune d’une pathologie neurodégénérative.2, 3
Le diagnostic positif repose sur l’identification d’antineuronaux dans le LCS et dans le sérum.2,7 Du fait d’un taux élevé de faux positifs, il est recommandé de faire confirmer tout test positif par le Centre de référence national (Lyon, Paris). Il existe d’exceptionnelles formes d’encéphalite sans anticorps détectable, dont le diagnostic repose sur des critères bien établis (tableau 3 ), en l’absence de diagnostic différentiel. Une tumeur sous-jacente doit être recherchée au minimum par un scanner thoraco-abdomino-pelvien et un TEP-scan.2 Selon l’anticorps mis en évidence, un bilan oncologique plus spécifique peut être nécessaire (tableaux 1 et 2 ).1, 2, 5
Le bilan comprend une IRM cérébrale, un EEG et une ponction lombaire. Si l’IRM révèle des hypersignaux hippocampiques, le principal diagnostic différentiel est le gliome, et le moindre doute sur un aspect infiltrant ou une prise de contraste impose une surveillance radioclinique rapprochée. Les examens biologiques doivent comporter une recherche £de troubles métaboliques et, selon le contexte, un bilan sérologique infectieux et un criblage des toxiques.2 La réalisation de PCR virales dans le liquide cérébrospinal (herpès simplex virus, virus d’Epstein-Barr, virus varicelle-zona) est primordiale. Enfin, l’évaluation des marqueurs de neurodégénérescence (Tau, phospho-Tau, protéine bêta-amyloïde) peut aider à distinguer une encéphalite auto-immune d’une pathologie neurodégénérative.2, 3
Le diagnostic positif repose sur l’identification d’antineuronaux dans le LCS et dans le sérum.2,7 Du fait d’un taux élevé de faux positifs, il est recommandé de faire confirmer tout test positif par le Centre de référence national (Lyon, Paris). Il existe d’exceptionnelles formes d’encéphalite sans anticorps détectable, dont le diagnostic repose sur des critères bien établis (
Un consensus d’experts pour le traitement
Le traitement de l’encéphalite auto-immune n’est pas strictement codifié et repose sur un consensus d’experts.1,3 En cas de forte suspicion diagnostique, un traitement de première ligne est mis en place le plus précocement possible, avant d’avoir obtenu le résultat de la recherche d’anticorps. On propose le plus souvent des corticoïdes (bolus intraveineux avec relais oral), généralement associés à des immunoglobulines intraveineuses. Les corticoïdes ne doivent cependant pas être débutés sans avoir auparavant vérifié, sur un scanner thoraco-abdomino-pelvien, qu’il n’y a pas d’adénomégalie compatible avec un lymphome. Une fois la confirmation diagnostique obtenue par la détection d’un anticorps spécifique, le traitement peut être intensifié. On propose le plus souvent en seconde ligne une association de rituximab et de cyclophosphamide intraveineux. La durée de traitement varie de six à dix-huit mois, selon l’évolution.1-3
En cas de forme paranéoplasique, les traitements de première ligne sont généralement maintenus tant qu’une chimiothérapie est nécessaire. À la fin du traitement oncologique, l’intensification du traitement immunosuppresseur doit être discutée.
Le traitement symptomatique doit quant à lui être adapté aux symptômes et à l’évolution. Par exemple, les antiépileptiques, souvent nécessaires à la phase d’état, peuvent généralement être arrêtés progressivement une fois le tableau bien stabilisé.
Une surveillance régulière est nécessaire au cours des premiers mois qui suivent le diagnostic, pour s’assurer de l’efficacité et de la tolérance du traitement. Il est préférable de faire un bilan semestriel de réévaluation complet (IRM cérébrale, EEG, ponction lombaire et bilan neuropsychologique) jusqu’à la stabilisation clinique. Ensuite, la surveillance est clinique, une fois par an.1-3,5,7
En cas de forme paranéoplasique, les traitements de première ligne sont généralement maintenus tant qu’une chimiothérapie est nécessaire. À la fin du traitement oncologique, l’intensification du traitement immunosuppresseur doit être discutée.
Le traitement symptomatique doit quant à lui être adapté aux symptômes et à l’évolution. Par exemple, les antiépileptiques, souvent nécessaires à la phase d’état, peuvent généralement être arrêtés progressivement une fois le tableau bien stabilisé.
Une surveillance régulière est nécessaire au cours des premiers mois qui suivent le diagnostic, pour s’assurer de l’efficacité et de la tolérance du traitement. Il est préférable de faire un bilan semestriel de réévaluation complet (IRM cérébrale, EEG, ponction lombaire et bilan neuropsychologique) jusqu’à la stabilisation clinique. Ensuite, la surveillance est clinique, une fois par an.1-3,5,7
Distinguer cibles de surface membranaire et intracellulaires
Les encéphalites auto-immunes sont des affections rares du système nerveux central. Elles se caractérisent par une atteinte neurologique aiguë ou subaiguë qui prédomine au niveau des régions limbiques, à l’origine d’une amnésie antérograde, d’une épilepsie temporale interne et de troubles psychiatriques. Elles sont associées à des anticorps antineuronaux ciblant des protéines de surface membranaire, le plus souvent non paranéoplasiques et en général de pronostic favorable, ou des protéines intracellulaires, souvent paranéoplasiques et de mauvais pronostic.
Un traitement immunomodulateur probabiliste peut être mis en place dès la suspicion clinique, après avoir éliminé les principaux diagnostics différentiels. Le pronostic semble sensiblement lié à la précocité d’introduction d’un traitement immunologique et à la prise en charge du cancer s’il existe. Les stratégies thérapeutiques sont encore peu codifiées, et ces recommandations sont destinées à évoluer en fonction des progrès de la recherche.
Un traitement immunomodulateur probabiliste peut être mis en place dès la suspicion clinique, après avoir éliminé les principaux diagnostics différentiels. Le pronostic semble sensiblement lié à la précocité d’introduction d’un traitement immunologique et à la prise en charge du cancer s’il existe. Les stratégies thérapeutiques sont encore peu codifiées, et ces recommandations sont destinées à évoluer en fonction des progrès de la recherche.
* Ondes delta de grande amplitude surmontées d’une activité thêta plus rapide.
Références
1. Graus F, Vogrig A, Muñiz-Castrillo S, Antoine JCG, Desestret V, Dubey D, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm 2021;8:e1014.
2. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391‑404.
3. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med 2018;378:840‑51.
4. Hébert J, Riche B, Vogrig A, Muñiz-Castrillo S, Joubert B, Picard G, et al. Epidemiology of paraneoplastic neurologic syndromes and autoimmune encephalitides in France. Neurol Neuroimmunol Neuroinflamm 2020;7:e883.
5. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157‑65.
6. Al-Diwani A, Handel A, Townsend L, Pollak T, Leite MI, Harrison PJ, et al. The psychopathology of NMDAR-antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry 2019;6:235‑46.
7. Ariño H, Armangué T, Petit-Pedrol M, Sabater L, Martinez-Hernandez E, Hara M, et al. Anti-LGI1-associated cognitive impairment: presentation and long-term outcome. Neurology 2016;87:759‑65.
8. Li W, Wu S, Meng Q, Zhang X, Guo Y, Cong L, et al. Clinical characteristics and short-term prognosis of LGI1 antibody encephalitis: a retrospective case study. BMC Neurol 2018;18:96.
9. Muñiz-Castrillo S, Joubert B, Elsensohn M-H, Pinto A-L, Saint-Martin M, Vogrig A, et al. Anti-CASPR2 clinical phenotypes correlate with HLA and immunological features. J Neurol Neurosurg Psychiatry 2020;91:1076‑84.
10. Joubert B, Belbezier A, Haesebaert J, Rheims S, Ducray F, Picard G, et al. Long-term outcomes in temporal lobe epilepsy with glutamate decarboxylase antibodies. J Neurol 2020;267:2083‑9.
2. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391‑404.
3. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med 2018;378:840‑51.
4. Hébert J, Riche B, Vogrig A, Muñiz-Castrillo S, Joubert B, Picard G, et al. Epidemiology of paraneoplastic neurologic syndromes and autoimmune encephalitides in France. Neurol Neuroimmunol Neuroinflamm 2020;7:e883.
5. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157‑65.
6. Al-Diwani A, Handel A, Townsend L, Pollak T, Leite MI, Harrison PJ, et al. The psychopathology of NMDAR-antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry 2019;6:235‑46.
7. Ariño H, Armangué T, Petit-Pedrol M, Sabater L, Martinez-Hernandez E, Hara M, et al. Anti-LGI1-associated cognitive impairment: presentation and long-term outcome. Neurology 2016;87:759‑65.
8. Li W, Wu S, Meng Q, Zhang X, Guo Y, Cong L, et al. Clinical characteristics and short-term prognosis of LGI1 antibody encephalitis: a retrospective case study. BMC Neurol 2018;18:96.
9. Muñiz-Castrillo S, Joubert B, Elsensohn M-H, Pinto A-L, Saint-Martin M, Vogrig A, et al. Anti-CASPR2 clinical phenotypes correlate with HLA and immunological features. J Neurol Neurosurg Psychiatry 2020;91:1076‑84.
10. Joubert B, Belbezier A, Haesebaert J, Rheims S, Ducray F, Picard G, et al. Long-term outcomes in temporal lobe epilepsy with glutamate decarboxylase antibodies. J Neurol 2020;267:2083‑9.
Dans cet article

Résumé
Les encéphalites auto-immunes sont des affections rares du système nerveux central liées à des anticorps antineuronaux. Leur présentation clinique classique associe amnésie antérograde, épilepsie temporale et troubles comportementaux, installés de façon subaiguë (en moins de trois mois). D’autres symptômes peuvent compléter le tableau, en fonction de l’anticorps. Les encéphalites auto-immunes par anticorps ciblant des protéines intracellulaires sont généralement paranéoplasiques et de mauvais pronostic. Celles qui sont associées à des anticorps ciblant des protéines de surface membranaire sont moins souvent contemporaines d’un cancer et de meilleur pronostic. Le diagnostic repose sur la mise en évidence de l’anticorps antineuronal. La prise en charge comporte le traitement d’une éventuelle néoplasie et des immunosuppresseurs.