La première administration d’une nouvelle molécule à des humains (volontaires « normaux » ou patients atteints d’un cancer en rechute) a pour but d’étudier sa pharmacologie, sa tolérance et sa sécurité d’emploi. Les règles de cette phase I sont très exigeantes et le risque zéro n’existe pas.
Le développement clinique des médicaments est précédé par des études réalisées chez l’animal et sur des modèles expérimentaux. Ces études précliniques sont résumées dans la brochure de l’investigateur et donnent des indications préliminaires sur les cibles pharmaco- logiques de la molécule en développement, ses caractéristiques pharmacocinétiques et les doses associées, chez l’animal, à diverses toxicités. L’interprétation de ces données non cliniques, enrichies au cours du développement de la molécule par les données cliniques au fur et à mesure qu’elles sont collectées, est complexe mais peut être facilitée par l’utilisation de logiciels d’analyse.1 Elle est cependant limitée par le fait que le meilleur modèle des humains reste l’humain. Les études cliniques sont donc indispensables pour vérifier que la molécule, à ce stade un produit médicamenteux en investigation, est suffisamment efficace et bien tolérée pour devenir un médicament utilisable dans la population cible dans la vie réelle. Cet article concerne les premières administrations d’une molécule à des humains, dites études de phase I du développement, en particulier l’étude de première administration à des humains.
Les différentes phases précoces de développement des médicaments
Les premières administrations d’un produit médicamenteux en investigation à des humains ont pour objectif d’étudier sa pharmacologie (pharmacocinétique et, si possible, pharmacodynamie), sa tolérance et sa sécurité d’emploi chez l’humain. Selon la nature du produit développé, ces études portent sur 20 à 150 volontaires. La toute première administration à des humains est particulièrement délicate dans la mesure où quasiment aucune information sur les données de sécurité dans l’espèce humaine, hors celles obtenues sur des modèles expérimentaux sur tissus ou cellules humaines, n’est disponible à ce stade. Récemment a été développé le concept d’études dites de phase 0, c’est-à-dire d’études de microdoses, pharmacologiquement inactives – et donc vraisemblablement inoffensives – mais permettant d’avoir une première idée du profil pharmacocinétique du produit en investigation et de son potentiel à provoquer des interactions médicamenteuses.2 L’exposition aux très faibles concentrations de ces phases 0 constitue cependant une limite à l’extrapolation de leurs résultats aux doses pharmacologiquement actives recherchées au cours des phases ultérieures de développement.
En dehors de la phase 0, non réglementaire et pas toujours réalisée, les études de phases précoces commencent par la première administration à des humains, appelée en anglais first-in-human study. Tout comme les autres études de phase I, elle est réalisée le plus souvent chez des sujets volontaires considérés comme « normaux ». Cependant, il y a des situations, notamment en oncologie, qui imposent l’inclusion de patients dans les études de phase I, y compris la première administration à des humains (v. infra). Les études de phase I ultérieures ont des objectifs variables qui dépendent des premières informations obtenues sur chaque molécule étudiée : précisions supplémentaires sur son profil cinétique et les possibles interactions médicamenteuses ; études spécifiques à différentes formes pharmaceutiques ; explorations pharma-codynamiques particulières, notamment sur ses effets sur la repolarisation ventriculaire cardiaque (un requis réglementaire systématique) ; études d’autres sources de variabilité cinétique ou dynamique, par exemple génétiques.
Enfin, hors du cadre de cet article, il est fréquent de réaliser des études chez des sujets « normaux » après la mise sur le marché d’un produit en investigation devenu un médicament approuvé par les autorités de santé. Ces études, improprement appelées de phase I puisque la phase I est par définition une phase réalisée avant commercialisation, souvent réalisées dans un cadre institutionnel, ont pour objectif d’étendre les connaissances sur un médicament, notamment pour étudier des interactions médicamenteuses, de nouvelles cibles pharmacodynamiques ou l’influence de polymorphismes génétiques sur la pharmacocinétique ou la pharmacodynamie. Des études de phase I de bioéquivalence sont également réalisées par les industriels pour la mise sur le marché de médicaments génériques.
En dehors de la phase 0, non réglementaire et pas toujours réalisée, les études de phases précoces commencent par la première administration à des humains, appelée en anglais first-in-human study. Tout comme les autres études de phase I, elle est réalisée le plus souvent chez des sujets volontaires considérés comme « normaux ». Cependant, il y a des situations, notamment en oncologie, qui imposent l’inclusion de patients dans les études de phase I, y compris la première administration à des humains (v. infra). Les études de phase I ultérieures ont des objectifs variables qui dépendent des premières informations obtenues sur chaque molécule étudiée : précisions supplémentaires sur son profil cinétique et les possibles interactions médicamenteuses ; études spécifiques à différentes formes pharmaceutiques ; explorations pharma-codynamiques particulières, notamment sur ses effets sur la repolarisation ventriculaire cardiaque (un requis réglementaire systématique) ; études d’autres sources de variabilité cinétique ou dynamique, par exemple génétiques.
Enfin, hors du cadre de cet article, il est fréquent de réaliser des études chez des sujets « normaux » après la mise sur le marché d’un produit en investigation devenu un médicament approuvé par les autorités de santé. Ces études, improprement appelées de phase I puisque la phase I est par définition une phase réalisée avant commercialisation, souvent réalisées dans un cadre institutionnel, ont pour objectif d’étendre les connaissances sur un médicament, notamment pour étudier des interactions médicamenteuses, de nouvelles cibles pharmacodynamiques ou l’influence de polymorphismes génétiques sur la pharmacocinétique ou la pharmacodynamie. Des études de phase I de bioéquivalence sont également réalisées par les industriels pour la mise sur le marché de médicaments génériques.
Aspects réglementaires
Il existe de très nombreuses recommandations officielles de l’Agence européenne du médicament sur le développement des médicaments,3 majoritairement harmonisées avec les recommandations japonaises et nord-américaines et adoptées par quasiment tous les pays du monde.* À la suite de l’accident dramatique survenu à Rennes** avec le BIA 10-2474,4, 5 les recommandations sur les études de première administration à des humains ont été mises à jour.6 Les recommandations pour le développement des anticancéreux sont en cours de révision.*** Le grand nombre de ces recommandations témoigne de l’extrême complexité du développement des médicaments et explique en partie l’augmentation du coût de ces développements et, indirectement, du prix des médicaments commercialisés. Il n’y a pas de modèle unique ou universel pour la réalisation des études de première administration à des humains ou celle des autres études de phase I. Chaque étude doit être adaptée au produit médicamenteux en investigation considéré en se référant aux recommandations officielles qui sont rédigées de manière volontairement ouverte et doivent donc être interprétées selon la nature du produit médicamenteux étudié. En France, la loi relative aux recherches impliquant la personne humaine (dite loi Jardé) a généré de nombreux décrets ciblant la gestion des événements et effets indésirables dans les études de première administration à des humains.
La toute première étude clinique d’administration à des humains7
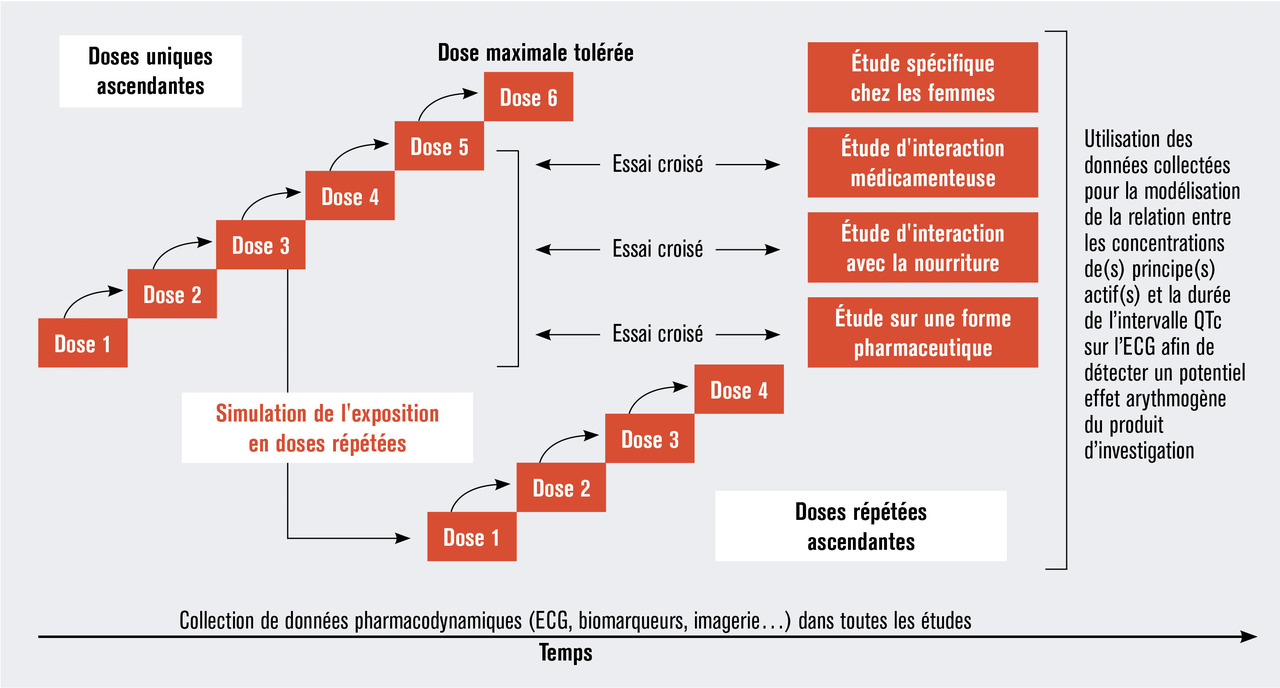
Les études de première administration à des humains débutent par l’administration de doses uniques ascendantes suivies, le plus souvent avant la fin de l’escalade des doses uniques, par celle de doses multiples (répétées) ascendantes à de petits groupes de volontaires, par exemple 6 volontaires dont 4 reçoivent le produit médicamenteux en investigation et 2 reçoivent son placebo en double insu (sauf en oncologie où tous les patients reçoivent le produit médicamenteux en investigation). La figure ci-contre illustre un plan expérimental communément utilisé pour une étude de première administration à des humains.
Le choix du niveau de la première dose est crucial pour la sécurité des volontaires. Il existe différentes méthodes de calcul de cette toute première dose, fondées sur des modèles d’extrapolation des données précliniques à l’humain. Les calculs sont différents pour les petites molécules et les produits de biotechnologie (notamment les anticorps thérapeutiques). L’utilisation d’un modèle animal approprié est donc essentielle, et l’exemple de l’accident survenu à Londres avec le TGN-1412**** illustre bien ce point.8 Les études précliniques du TGN-1412, un anticorps agoniste du récepteur CD28 activant les lymphocytes T via son corécepteur CD4 entraînant ainsi la libération de cytokines, ont été réalisées chez des singes macaques. Or cette espèce animale perd son récepteur CD28 dès l’activation de CD4 et est donc insensible à un effet agoniste CD28. Le TGN 1412 a pu être redéveloppé chez les humains en débutant par l’administration de doses 100 fois inférieures à celles ayant provoqué l’accident à Londres.9 Typiquement, pour les petites molécules, on calcule une dose à partir de la dose la plus élevée n’ayant pas provoqué d’effet indésirable dans l’espèce animale la plus sensible convertie à l’espèce humaine.7 Pour les anticorps, le calcul se fonde en général sur la dose la plus faible ayant eu un effet biologique sur un marqueur d’activité. D’autres méthodes existent.7
Le choix de la progression des doses dépend des risques d’effets indésirables, estimés puis observés avec le produit en développement, et de son profil pharmacocinétique obtenu avec les premières doses administrées. Ce choix est un compromis entre la nécessaire – et prioritaire – garantie de la sécurité des volontaires participant à l’étude et la nécessité d’explorer des doses pharmacologiquement actives susceptibles d’être étudiées dans les phases ultérieures de développement avec le risque d’abandonner le développement d’une molécule thérapeutiquement utile faute d’avoir su augmenter les doses testées. Après la première dose, il est habituel d’administrer une dose deux fois supérieure à chaque nouveau palier de dose, du moins pour les premiers d’entre eux. Des incréments plus faibles sont aussi utilisés, par exemple selon une suite de Fibonacci dans laquelle chaque incrément de dose est la somme des deux doses précédemment administrées (1, 1, 2, 3, 5, 8, 13, 21, 34…). L’incrémentation est également fondée sur l’analyse des données pharmacocinétiques à chaque palier de dose. S’il existe un doute sur la non-linéarité de la cinétique à un palier de dose (les concentrations ont augmenté plus que de manière proportionnelle à l’augmentation de la dose), l’augmentation de la dose au palier suivant est plus faible. Mais, surtout, la progression des doses est déterminée par les données cliniques, et dans une moindre mesure par les données biologiques, de tolérance.
Le choix de l’arrêt de l’augmentation des doses, et de l’arrêt de l’étude, est variable selon les produit médicamenteux en investigation.10 La survenue, à une dose donnée, d’un effet indésirable grave chez un seul sujet, ou d’un effet indésirable d’intensité élevée même non grave chez deux sujets, possiblement liés au produit médicamenteux en investigation, constitue un critère d’arrêt de l’augmentation des doses, voire de l’étude.6 D’autres critères plus exigeants peuvent être utilisés, seuls, combinés, et/ou en association avec des critères biologiques pour décider d’arrêter d’augmenter les doses. Là encore, il n’y a pas de règle universelle, et chaque promoteur propose ses critères aux autorités de régulation (Agence du médicament) et au comité de protection des personnes examinant le protocole, pour chaque étude, avant et en cours d’étude. Depuis l’accident survenu à Rennes en 2016, il est également suggéré d’arrêter d’augmenter les doses lorsque la cible pharmacologique a été atteinte, mais ce critère est souvent difficile à définir précisément.6
Le choix du niveau de la première dose est crucial pour la sécurité des volontaires. Il existe différentes méthodes de calcul de cette toute première dose, fondées sur des modèles d’extrapolation des données précliniques à l’humain. Les calculs sont différents pour les petites molécules et les produits de biotechnologie (notamment les anticorps thérapeutiques). L’utilisation d’un modèle animal approprié est donc essentielle, et l’exemple de l’accident survenu à Londres avec le TGN-1412**** illustre bien ce point.8 Les études précliniques du TGN-1412, un anticorps agoniste du récepteur CD28 activant les lymphocytes T via son corécepteur CD4 entraînant ainsi la libération de cytokines, ont été réalisées chez des singes macaques. Or cette espèce animale perd son récepteur CD28 dès l’activation de CD4 et est donc insensible à un effet agoniste CD28. Le TGN 1412 a pu être redéveloppé chez les humains en débutant par l’administration de doses 100 fois inférieures à celles ayant provoqué l’accident à Londres.9 Typiquement, pour les petites molécules, on calcule une dose à partir de la dose la plus élevée n’ayant pas provoqué d’effet indésirable dans l’espèce animale la plus sensible convertie à l’espèce humaine.7 Pour les anticorps, le calcul se fonde en général sur la dose la plus faible ayant eu un effet biologique sur un marqueur d’activité. D’autres méthodes existent.7
Le choix de la progression des doses dépend des risques d’effets indésirables, estimés puis observés avec le produit en développement, et de son profil pharmacocinétique obtenu avec les premières doses administrées. Ce choix est un compromis entre la nécessaire – et prioritaire – garantie de la sécurité des volontaires participant à l’étude et la nécessité d’explorer des doses pharmacologiquement actives susceptibles d’être étudiées dans les phases ultérieures de développement avec le risque d’abandonner le développement d’une molécule thérapeutiquement utile faute d’avoir su augmenter les doses testées. Après la première dose, il est habituel d’administrer une dose deux fois supérieure à chaque nouveau palier de dose, du moins pour les premiers d’entre eux. Des incréments plus faibles sont aussi utilisés, par exemple selon une suite de Fibonacci dans laquelle chaque incrément de dose est la somme des deux doses précédemment administrées (1, 1, 2, 3, 5, 8, 13, 21, 34…). L’incrémentation est également fondée sur l’analyse des données pharmacocinétiques à chaque palier de dose. S’il existe un doute sur la non-linéarité de la cinétique à un palier de dose (les concentrations ont augmenté plus que de manière proportionnelle à l’augmentation de la dose), l’augmentation de la dose au palier suivant est plus faible. Mais, surtout, la progression des doses est déterminée par les données cliniques, et dans une moindre mesure par les données biologiques, de tolérance.
Le choix de l’arrêt de l’augmentation des doses, et de l’arrêt de l’étude, est variable selon les produit médicamenteux en investigation.10 La survenue, à une dose donnée, d’un effet indésirable grave chez un seul sujet, ou d’un effet indésirable d’intensité élevée même non grave chez deux sujets, possiblement liés au produit médicamenteux en investigation, constitue un critère d’arrêt de l’augmentation des doses, voire de l’étude.6 D’autres critères plus exigeants peuvent être utilisés, seuls, combinés, et/ou en association avec des critères biologiques pour décider d’arrêter d’augmenter les doses. Là encore, il n’y a pas de règle universelle, et chaque promoteur propose ses critères aux autorités de régulation (Agence du médicament) et au comité de protection des personnes examinant le protocole, pour chaque étude, avant et en cours d’étude. Depuis l’accident survenu à Rennes en 2016, il est également suggéré d’arrêter d’augmenter les doses lorsque la cible pharmacologique a été atteinte, mais ce critère est souvent difficile à définir précisément.6
Sélection des participants
Les études de phase précoce du développement des médicaments sont réalisées par des investigateurs et des équipes spécialisées et se déroulent dans des centres expérimentés dans leur réalisation. En France, les agences régionales de santé (ARS) délivrent des autorisations de lieu pour la réalisation de ces études, pour un site inspecté par l’ARS de la région et pour un responsable qualifié. La sélection des volontaires pour les études de phase I repose sur la véri- fication de la « normalité » de leurs caractéristiques cliniques et bio- logiques de routine (fonction rénale et hépatique, hémogramme, tests virologiques, entre autres). Il est important de garder en tête que la normalité ne peut être que relative dans la mesure où on ne peut définir de manière parfaite ce qu’est un sujet sain. D’ailleurs, le terme « sain » n’apparaît dans aucun texte réglementaire. Les critères de sélection sont relativement standardisés10 avec cette limite, notamment pour les études de première administration à l’homme, que toutes les caractéristiques bio- logiques d’un humain ne peuvent être connues. Les exigences de normalité peuvent varier selon le produit étudié, notamment par la sélection de critères spécifiques ciblant la pharmacodynamie de la molécule développée. Les critères de normalité peuvent être sujets à interprétation de la part des investigateurs, sachant que des variations faibles de la « normalité » existent chez 2 à 5 %, voire 10 % pour certains paramètres, des sujets recevant un placebo.11, 12
Les investigateurs sont attentifs au fait que la motivation première des volontaires « normaux » pour participer à une étude est l’indemnisation financière que la loi prévoit pour leur participation (jusqu’à 4 500 € par an). La transparence de part et d’autre s’impose. La loi française est protectrice du fait de l’existence d’un fichier national des volontaires qui permet de vérifier qu’un sujet donné n’a pas récemment participé à une autre étude qui le disqualifierait pour être inclus dans une nouvelle étude, soit qu’il ait atteint le maximum d’indemnisation autorisé par la loi, soit qu’il soit encore dans une période d’exclusion suivant sa participation à une étude antérieure.
Les investigateurs sont attentifs au fait que la motivation première des volontaires « normaux » pour participer à une étude est l’indemnisation financière que la loi prévoit pour leur participation (jusqu’à 4 500 € par an). La transparence de part et d’autre s’impose. La loi française est protectrice du fait de l’existence d’un fichier national des volontaires qui permet de vérifier qu’un sujet donné n’a pas récemment participé à une autre étude qui le disqualifierait pour être inclus dans une nouvelle étude, soit qu’il ait atteint le maximum d’indemnisation autorisé par la loi, soit qu’il soit encore dans une période d’exclusion suivant sa participation à une étude antérieure.
Le cas particulier de l’oncologie
Les études de pharmacologie précoce en oncologie sont particulières en ce sens que les malades qui y participent sont, dans la majorité des cas, des patients ayant un cancer réfractaire à d’autres traitements oncologiques et qui espèrent un bénéfice clinique d’un nouveau traitement expérimental. Ces études de phase précoce en oncologie laissaient peu d’espoir de guérison ou de rémission avec les molécules cytotoxiques dans les années 1990. Les études plus récentes de phase précoce durant ces cinq dernières années, utilisant des produits innovants de biotechnologie ou des inhibiteurs de protéines kinases, ont montré des réponses complètes ou partielles de l’ordre de 20 %.13 Un tel taux de réponse dans une population en échec thérapeutique à l’inclusion est excellent et témoigne des progrès actuels réalisés en oncologie. Les développements de médicaments en oncologie ont également la particularité de souvent associer étroitement les phases précoces aux phases ultérieures : l’étude de première administration à des humains inclut souvent des patients ayant des cancers de différents types ; selon les réponses obtenues en phase précoce et la meilleure caractérisation des patients répondeurs (notamment par des marqueurs tumoraux de réponse), le développement se poursuit, parfois chez les mêmes patients, de manière plus ciblée.
Données de sécurité
Des analyses récentes des études de phase I non oncologiques portant sur plusieurs milliers de sujets « normaux » indiquent que ces études ne sont associées qu’à des effets indésirables mineurs et à un taux très faible d’événements indésirables graves, de l’ordre de 0,3 %.14, 15
Les recommandations officielles, qui ont été mises à jour après l’accident survenu à Rennes avec le BIA 10-2474, ont intensifié le niveau des exigences sécuritaires des études de phase précoce, pourtant déjà élevé auparavant.6 Elles prévoient notamment que la première administration de la première dose ne soit délivrée qu’à un seul volontaire, dit sujet « sentinelle », ou à deux sujets dont l’un reçoit le placebo. Le sujet sentinelle est surveillé, cliniquement et biologiquement, suffisamment longtemps pour poursuivre l’étude chez d’autres volontaires. Ce principe s’applique à la phase des doses uniques comme à celle des doses répétées des études de première administration à des humains. À chaque palier de dose, les données cliniques de tolérance et les données pharmacocinétiques doivent être analysées avant de passer à la dose suivante, quitte à la modifier à la baisse. Le promoteur de l’étude doit informer « sans délai » les autorités de santé de tout événement indésirable grave.
Mais au-delà des recommandations officielles, c’est d’abord aux investigateurs que revient la responsabilité décisionnelle de poursuivre la participation d’un volontaire, l’escalade des doses et l’étude elle-même.5 Et la première règle à suivre est de considérer que tout événement indésirable grave, dont la définition est indiquée dans letableau 1 , est jusqu’à preuve du contraire, lié à l’administration du produit médicamenteux en investigation.
Enfin, du fait du faible nombre de sujets étudiés au cours des phases précoces de développement des médicaments, l’absence de survenue d’un effet indésirable grave au cours de ces premières études ne garantit pas qu’un tel effet indésirable grave sera absent des phases de développement ultérieures, voire de son utilisation après commercialisation. Le tableau 2 illustre ce point de simple probabilité.
Les recommandations officielles, qui ont été mises à jour après l’accident survenu à Rennes avec le BIA 10-2474, ont intensifié le niveau des exigences sécuritaires des études de phase précoce, pourtant déjà élevé auparavant.6 Elles prévoient notamment que la première administration de la première dose ne soit délivrée qu’à un seul volontaire, dit sujet « sentinelle », ou à deux sujets dont l’un reçoit le placebo. Le sujet sentinelle est surveillé, cliniquement et biologiquement, suffisamment longtemps pour poursuivre l’étude chez d’autres volontaires. Ce principe s’applique à la phase des doses uniques comme à celle des doses répétées des études de première administration à des humains. À chaque palier de dose, les données cliniques de tolérance et les données pharmacocinétiques doivent être analysées avant de passer à la dose suivante, quitte à la modifier à la baisse. Le promoteur de l’étude doit informer « sans délai » les autorités de santé de tout événement indésirable grave.
Mais au-delà des recommandations officielles, c’est d’abord aux investigateurs que revient la responsabilité décisionnelle de poursuivre la participation d’un volontaire, l’escalade des doses et l’étude elle-même.5 Et la première règle à suivre est de considérer que tout événement indésirable grave, dont la définition est indiquée dans le
Enfin, du fait du faible nombre de sujets étudiés au cours des phases précoces de développement des médicaments, l’absence de survenue d’un effet indésirable grave au cours de ces premières études ne garantit pas qu’un tel effet indésirable grave sera absent des phases de développement ultérieures, voire de son utilisation après commercialisation. Le
PAS DE RISQUE zéro
Les études de phase précoce du développement des médicaments sont complexes et très diversifiées dans leurs plans d’expérience. Leur encadrement réglementaire est très exigeant, tant pour les conditions dans lesquelles elles sont réalisées que pour la qualification des investigateurs et des équipes les réalisant. Ces exigences expliquent que ces études ne sont qu’exceptionnellement responsables d’effets indésirables graves et encore moins souvent de décès. L’accident de Londres avec le TGN 14128 et de Rennes avec le BIA 10-24745 nous rappellent cependant que le risque zéro n’existe pas.
* www.ich.org/
** Le BIA 10-2474 est un inhibiteur d’une enzyme qui métabolise les endocannabinoïdes. Alors que 84 volon- taires sains avaient reçu la molécule en doses uniques ou multiples ascendantes ou lors d’une étude d'interaction avec la nourriture sans signal d’intolérance, 5 des 6 sujets recevant la 5e de 10 doses du 5e groupe recevant les doses multiples ont développé des effets indésirables neurologiques graves liés à des micro- hémorragies. Un de ces volontaires est décédé de ces lésions cérébrales. Le mécanisme exact de cet accident reste inconnu.
*** www.ema.europa.eu/en/evaluation-anticancer-medicinal-products-man**** Le TGN-1412 est un immunomodulateur qui, lors de sa première administration à des volontaires sains dans un centre spécialisé à Londres en 2006, a provoqué de graves réactions inflammatoires dues à une libération massive de cytokines. La dose choisie pour cette première administration s’est révélée beaucoup trop élevée. Le produit a pu être redéveloppé à une dose beaucoup plus faible, sous le nom de thérazumab. Lors de l’accident, le TGN-1412 a été administré « à la chaîne » chez 6 sujets sains alors même que le premier volontaire développait immédiatement de graves symptômes.
** Le BIA 10-2474 est un inhibiteur d’une enzyme qui métabolise les endocannabinoïdes. Alors que 84 volon- taires sains avaient reçu la molécule en doses uniques ou multiples ascendantes ou lors d’une étude d'interaction avec la nourriture sans signal d’intolérance, 5 des 6 sujets recevant la 5e de 10 doses du 5e groupe recevant les doses multiples ont développé des effets indésirables neurologiques graves liés à des micro- hémorragies. Un de ces volontaires est décédé de ces lésions cérébrales. Le mécanisme exact de cet accident reste inconnu.
*** www.ema.europa.eu/en/evaluation-anticancer-medicinal-products-man**** Le TGN-1412 est un immunomodulateur qui, lors de sa première administration à des volontaires sains dans un centre spécialisé à Londres en 2006, a provoqué de graves réactions inflammatoires dues à une libération massive de cytokines. La dose choisie pour cette première administration s’est révélée beaucoup trop élevée. Le produit a pu être redéveloppé à une dose beaucoup plus faible, sous le nom de thérazumab. Lors de l’accident, le TGN-1412 a été administré « à la chaîne » chez 6 sujets sains alors même que le premier volontaire développait immédiatement de graves symptômes.
Références
1. Van Gerven J, Cohen A. Integrating data from the Investigational Medicinal Product Dossier/investigator's brochure. A new tool for translational integration of preclinical effects. Br J Clin Pharmacol 2018;84:1457-66.
2. Burt T, Yoshida K, Lappin G, et al. Microdosing and other phase 0 clinical trials: facilitating translation in drug development. Clin Transl Sci 2016;9:74-88.
3. European Medicines Agency. Research and development. Recommandations EMA pour la recherche et le développement. https://www.ema.europa.eu/en/human-regulatory/research-development
4. Ménard J. Drame de Rennes : que s'est-il passé ? Quelles en sont les conséquences pour les essais futurs ? Rev Prat 2016;66:481-3.
5. Funck-Brentano C, Ménard J. The BIAL/Biotrial case of death of a human volunteer in the first-in-human study of BIA 10-2474: Are we missing the fundamentals? Presse Med 2016;45:719-22.
6. European Medicines Agency Strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. EMEA/CHMP/SWP/28367/07 Rev. 1, 2017. https://www.ema.europa.eu/en/strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational-medicinal
7. Shen J, Swift B, Mamelok R, Pine S, Sinclair J, Attar M. Design and conduct considerations for first-in-human trials. Clin Transl Sci 2019;12:6-19.
8. Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006;355:1018-28.
9. Kenter MJ, Cohen AF. The return of the prodigal son and the extraordinary development route of antibody TGN1412 - lessons for drug development and clinical pharmacology. Br J Clin Pharmacol 2015;79:545-7.
10. Breithaupt-Groegler K, Coch C, Coenen M, et al. Who is a 'healthy subject'?-consensus results on pivotal eligibility criteria for clinical trials. Eur J Clin Pharmacol 2017;73:409-16.
11. Clayton GL, Schachter AD, Magnusson B, Li Y, Colin L. How often do safety signals occur by chance in first-in-human trials? Clin Transl Sci 2018;11:471-6.
12. Young TC, Srinivasan S, Vetter ML, et al. A systematic review and pooled analysis of select safety parameters among normal healthy volunteers taking placebo in phase 1 clinical trials. J Clin Pharmacol 2017;57:1079-87.
13. Chakiba C, Grellety T, Bellera C, Italiano A. Encouraging trends in modern phase 1 oncology trials. N Engl J Med 2018;378:2242-3.
14. Johnson RA, Rid A, Emanuel E, Wendler D. Risks of phase I research with healthy participants: a systematic review. Clin Trials 2016;13:149-60.
15. Emanuel EJ, Bedarida G, Macci K, Gabler NB, Rid A, Wendler D. Quantifying the risks of non-oncology phase I research in healthy volunteers: meta-analysis of phase I studies. BMJ 2015;350:h3271.
2. Burt T, Yoshida K, Lappin G, et al. Microdosing and other phase 0 clinical trials: facilitating translation in drug development. Clin Transl Sci 2016;9:74-88.
3. European Medicines Agency. Research and development. Recommandations EMA pour la recherche et le développement. https://www.ema.europa.eu/en/human-regulatory/research-development
4. Ménard J. Drame de Rennes : que s'est-il passé ? Quelles en sont les conséquences pour les essais futurs ? Rev Prat 2016;66:481-3.
5. Funck-Brentano C, Ménard J. The BIAL/Biotrial case of death of a human volunteer in the first-in-human study of BIA 10-2474: Are we missing the fundamentals? Presse Med 2016;45:719-22.
6. European Medicines Agency Strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. EMEA/CHMP/SWP/28367/07 Rev. 1, 2017. https://www.ema.europa.eu/en/strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational-medicinal
7. Shen J, Swift B, Mamelok R, Pine S, Sinclair J, Attar M. Design and conduct considerations for first-in-human trials. Clin Transl Sci 2019;12:6-19.
8. Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006;355:1018-28.
9. Kenter MJ, Cohen AF. The return of the prodigal son and the extraordinary development route of antibody TGN1412 - lessons for drug development and clinical pharmacology. Br J Clin Pharmacol 2015;79:545-7.
10. Breithaupt-Groegler K, Coch C, Coenen M, et al. Who is a 'healthy subject'?-consensus results on pivotal eligibility criteria for clinical trials. Eur J Clin Pharmacol 2017;73:409-16.
11. Clayton GL, Schachter AD, Magnusson B, Li Y, Colin L. How often do safety signals occur by chance in first-in-human trials? Clin Transl Sci 2018;11:471-6.
12. Young TC, Srinivasan S, Vetter ML, et al. A systematic review and pooled analysis of select safety parameters among normal healthy volunteers taking placebo in phase 1 clinical trials. J Clin Pharmacol 2017;57:1079-87.
13. Chakiba C, Grellety T, Bellera C, Italiano A. Encouraging trends in modern phase 1 oncology trials. N Engl J Med 2018;378:2242-3.
14. Johnson RA, Rid A, Emanuel E, Wendler D. Risks of phase I research with healthy participants: a systematic review. Clin Trials 2016;13:149-60.
15. Emanuel EJ, Bedarida G, Macci K, Gabler NB, Rid A, Wendler D. Quantifying the risks of non-oncology phase I research in healthy volunteers: meta-analysis of phase I studies. BMJ 2015;350:h3271.
Dans cet article

Résumé
Les premières administrations d’une molécule à des humains, dites études de phase I du développement font suite aux études expérimentales qui permettent d’avoir une première idée du profil de ses effets pharmacologiques et de sa toxicité. Ces études sont typiquement organisées avec des sujets « normaux » ou, en oncologie, chez des patients en rechute. La sécurité des participants est une priorité. Des doses uniques, suivies de doses répétées, sont administrées par des professionnels formés dans des lieux médicalisés autorisés. Elles permettent d’étudier le profil pharmacocinétique et pharmacodynamique de la molécule chez les humains et d’explorer quelques sources de variabilité de ces paramètres. Elles sont très encadrées réglementairement et relativement stéréotypées méthodologiquement.