L’attribution du prix Nobel de médecine à James Allison de l’université du Texas et à Tasuku Honjo de l’université de Kyoto récompense des années de travaux expérimentaux sur les mécanismes de contrôle de la réponse immunitaire, ayant permis de la reprogrammer afin de vaincre le cancer.
En validant l’efficacité de l’immunothérapie, James Allison et Tasuku Honjo (fig. 1) ont ouvert la voie à l’élaboration d’une nouvelle classe d’agents anticancéreux, les inhibiteurs du contrôle immunitaire (immune checkpoint inhibitors [ICI]) et à une nouvelle conception des traitements des cancers associant les traitements classiques (chirurgie, radiothérapie, chimiothérapie) et l’immunothérapie.
Réponse immunitaire anticancéreuse
Cette révolution médicale a remis en lumière la notion de réponse immunitaire anticancéreuse, suspectée dès la fin du XIXe siècle par William B. Cooley, qui a rapporté des cas de régression de tumeur par l’injection d’extraits bactériens, mais surtout démontrée par Georges Mathé il y a plus de 50 ans par la guérison d’une leucémie chez un homme grâce à la greffe de moelle osseuse de son jumeau.1
Les nombreuses tentatives dans les années 1970-1980 pour « stimuler »la réponse immunitaire anticancéreuse n’ont pas connu le succès escompté en dehors de l’utilisation du BCG dans le cancer de la vessie, en raison de l’absence de connaissances suffisantes sur les mécanismes de fonctionnement du système immunitaire. Les tentatives d’immunostimulation reposaient essentiellement sur l’utilisation de molécules chimiques ou d’extraits bactériens qui devaient être des agonistes des récepteurs TOLL à une époque où ceux-ci n’avaient pas encore été décrits par Jules Hoffmann, Prix Nobel de médecine en 2011.2
La mise en évidence de cellules immunitaires au sein des tumeurs a conduit Steven Rosenberg à prélever les lymphocytes T des patients, les activer et les expandre in vitro avec de l’interleukine 2, avant de les réinjecter.3 La démonstration que ces tumor infiltrating lymphocytes (TIL) étaient capables d’entraîner la régression de tumeurs a apporté un élément supplémentaire dans la validation d’une réponse immunitaire anticancéreuse efficace et dans la possibilité de traiter les cancers par l’immunothérapie.
Les interactions entre cellules immunitaires, cellules tumorales et micro-environnement ont alors été théorisées par Robert Schneiber sous la règle des trois « é » : élimination des cellules tumorales par les cellules immunitaires, équilibre (maîtrise de la prolifération des cellules tumorales sans éradication) puis échappement, seule phase visible.4
Les nombreuses tentatives dans les années 1970-1980 pour « stimuler »la réponse immunitaire anticancéreuse n’ont pas connu le succès escompté en dehors de l’utilisation du BCG dans le cancer de la vessie, en raison de l’absence de connaissances suffisantes sur les mécanismes de fonctionnement du système immunitaire. Les tentatives d’immunostimulation reposaient essentiellement sur l’utilisation de molécules chimiques ou d’extraits bactériens qui devaient être des agonistes des récepteurs TOLL à une époque où ceux-ci n’avaient pas encore été décrits par Jules Hoffmann, Prix Nobel de médecine en 2011.2
La mise en évidence de cellules immunitaires au sein des tumeurs a conduit Steven Rosenberg à prélever les lymphocytes T des patients, les activer et les expandre in vitro avec de l’interleukine 2, avant de les réinjecter.3 La démonstration que ces tumor infiltrating lymphocytes (TIL) étaient capables d’entraîner la régression de tumeurs a apporté un élément supplémentaire dans la validation d’une réponse immunitaire anticancéreuse efficace et dans la possibilité de traiter les cancers par l’immunothérapie.
Les interactions entre cellules immunitaires, cellules tumorales et micro-environnement ont alors été théorisées par Robert Schneiber sous la règle des trois « é » : élimination des cellules tumorales par les cellules immunitaires, équilibre (maîtrise de la prolifération des cellules tumorales sans éradication) puis échappement, seule phase visible.4
Activation des lymphocytes T
L’activation et la prolifération des lymphocytes T naïfs nécessitent deux signaux intracellulaires, un premier signal lié à la reconnaissance de l’antigène par le récepteur T spécifique et un second signal dit de costimulation. Dès 1990, Ron Schwartz avait souligné l’importance de ce second signal.5 En effet, l’engagement du premier signal, sans le second, rend la cellule anergique, c’est-à-dire incapable de s’activer et de proliférer, même quand elle est stimulée ultérieurement par les deux signaux.
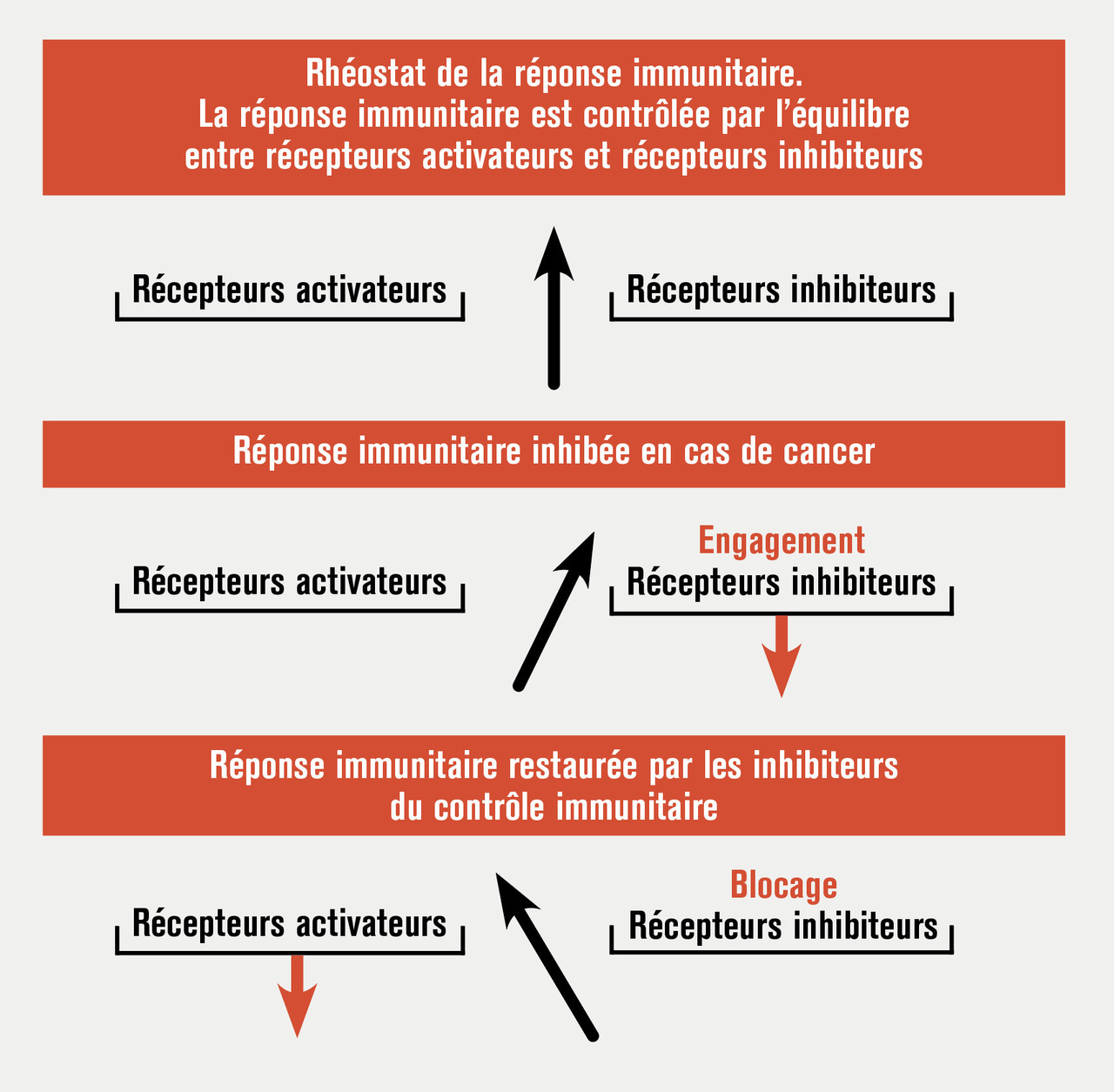
La production des anticorps monoclonaux, dont la découverte a valu à Kohler et Milstein l’attribution du prix Nobel de médecine en 1984, a permis de disséquer les antigènes membranaires des lymphocytes T et de mieux comprendre la physiologie de ces cellules. La principale molécule de costimulation des lymphocytes T, CD28, interagit avec les molécules B7, B7-1 (CD80) et B7-2 (CD86) des cellules présentatrices d’antigène pour induire l’activation, la prolifération et la survie des lymphocytes T.6 Très rapidement, il s’est avéré que ce rhéostat était plus complexe et comportait l’implication de récepteurs activateurs des lymphocytes T permettant d’amplifier la réponse immunitaire pour éliminer rapidement un pathogène (CD28, 4-1BB, ICOS...) et de récepteurs inhibiteurs (CTLA-4 [cytotoxic T lymphocyte associated proteine-4], PD-1 [programmed cell death-1]) pour la contrôler une fois le pathogène éliminé. C’est de cet équilibre subtil entre récepteurs activateurs et récepteurs inhibiteurs que dépend toute réponse immunitaire.
C’est en 1994 que l’activité inhibitrice de CTLA-4, molécule découverte par le Français Pierre Golstein en 1987, a été caractérisée.7 En 1995, James Allison a montré que CTLA-4, qui présente 80 % d’homologie avec CD28, avait les mêmes ligands que CD28, c’est-à-dire B7-1 (CD80) et B7-2 (CD86), mais avec une fonction opposée (inhibitrice au lieu d’activatrice).8 CTLA-4, contrairement à CD28, n’est pas exprimé de façon constitutive à la membrane des lymphocytes T naïfs mais apparaît rapidement chez les lymphocytes T activés et les lymphocytes T régulateurs. Comme son affinité pour les molécules B7 est beaucoup plus importante, la liaison CTLA 4-B7 (inhibitrice) se substitue à la liaison CD28-B7, réalisant ainsi une inhibition par feed back des lymphocytes T activés, comme l’a démontré la survenue de prolifération fatale de lymphocytes T chez les souris dont le gène pour CTLA-4 a été inactivé (CTL4-4 KO).9 Cette affinité de CTLA-4 pour les molécules B7 a été utilisée pour construire des protéines de fusion, associant le fragment de CTLA-4 qui se lie à B7 avec le fragment fixe d’une immunoglobuline (CTLA4-Ig), pour inhiber la liaison entre B7 et CD28 et bloquer le signal activateur de costimulation. Ces protéines de fusion (abatacept et bélatacept) sont utilisées comme immunosuppresseurs dans le traitement des maladies inflammatoires rhumatismales et pour prévenir le rejet de greffe d’organes.
La production des anticorps monoclonaux, dont la découverte a valu à Kohler et Milstein l’attribution du prix Nobel de médecine en 1984, a permis de disséquer les antigènes membranaires des lymphocytes T et de mieux comprendre la physiologie de ces cellules. La principale molécule de costimulation des lymphocytes T, CD28, interagit avec les molécules B7, B7-1 (CD80) et B7-2 (CD86) des cellules présentatrices d’antigène pour induire l’activation, la prolifération et la survie des lymphocytes T.6 Très rapidement, il s’est avéré que ce rhéostat était plus complexe et comportait l’implication de récepteurs activateurs des lymphocytes T permettant d’amplifier la réponse immunitaire pour éliminer rapidement un pathogène (CD28, 4-1BB, ICOS...) et de récepteurs inhibiteurs (CTLA-4 [cytotoxic T lymphocyte associated proteine-4], PD-1 [programmed cell death-1]) pour la contrôler une fois le pathogène éliminé. C’est de cet équilibre subtil entre récepteurs activateurs et récepteurs inhibiteurs que dépend toute réponse immunitaire.
C’est en 1994 que l’activité inhibitrice de CTLA-4, molécule découverte par le Français Pierre Golstein en 1987, a été caractérisée.7 En 1995, James Allison a montré que CTLA-4, qui présente 80 % d’homologie avec CD28, avait les mêmes ligands que CD28, c’est-à-dire B7-1 (CD80) et B7-2 (CD86), mais avec une fonction opposée (inhibitrice au lieu d’activatrice).8 CTLA-4, contrairement à CD28, n’est pas exprimé de façon constitutive à la membrane des lymphocytes T naïfs mais apparaît rapidement chez les lymphocytes T activés et les lymphocytes T régulateurs. Comme son affinité pour les molécules B7 est beaucoup plus importante, la liaison CTLA 4-B7 (inhibitrice) se substitue à la liaison CD28-B7, réalisant ainsi une inhibition par feed back des lymphocytes T activés, comme l’a démontré la survenue de prolifération fatale de lymphocytes T chez les souris dont le gène pour CTLA-4 a été inactivé (CTL4-4 KO).9 Cette affinité de CTLA-4 pour les molécules B7 a été utilisée pour construire des protéines de fusion, associant le fragment de CTLA-4 qui se lie à B7 avec le fragment fixe d’une immunoglobuline (CTLA4-Ig), pour inhiber la liaison entre B7 et CD28 et bloquer le signal activateur de costimulation. Ces protéines de fusion (abatacept et bélatacept) sont utilisées comme immunosuppresseurs dans le traitement des maladies inflammatoires rhumatismales et pour prévenir le rejet de greffe d’organes.
Inhibiteurs du contrôle immunitaire (ICI)
La réponse immunitaire étant contrôlée par un équilibre subtil entre récepteurs activateurs et récepteurs inhibiteurs, considérant que l’environnement tumoral très immunosuppresseur favorisait l’engagement des récepteurs inhibiteurs, James Allison a eu l’intuition géniale de bloquer l’action du récepteur inhibiteur en utilisant un anticorps anti-CTLA-4 pour rétablir une liaison activatrice B7-CD28 et libérer la réponse immunitaire de son frein (fig. 2). Il a apporté en 1996 dans Science la preuve du concept de l’efficacité de cette nouvelle immunothérapie en démontrant dans un modèle murin que l’administration d’un anti-CTLA-4 pouvait traiter des tumeurs.10
Cette découverte a bien sûr suscité l’utilisation des anticorps anti-CTLA-4 chez l’homme. Un premier anticorps, l’ipilimumab, a été utilisé dans le traitement de cancers, et des résultats favorables dans 20 % des cas de mélanomes métastatiques ont été rapportés en 2010,11 justifiant sa reconnaissance comme premier inhibiteur du contrôle immunitaire (ICI), par la Food and Drug Administration (FDA) en 2011.Des travaux ultérieurs ont confirmé cette étude initiale, les résultats poolés dans le mélanome de stade III ou IV montrant chez 20 % des patients une réponse clinique durable avec un plateau débutant aux alentours de la troisième année, résultats bien supérieurs à ce qui était observé auparavant.
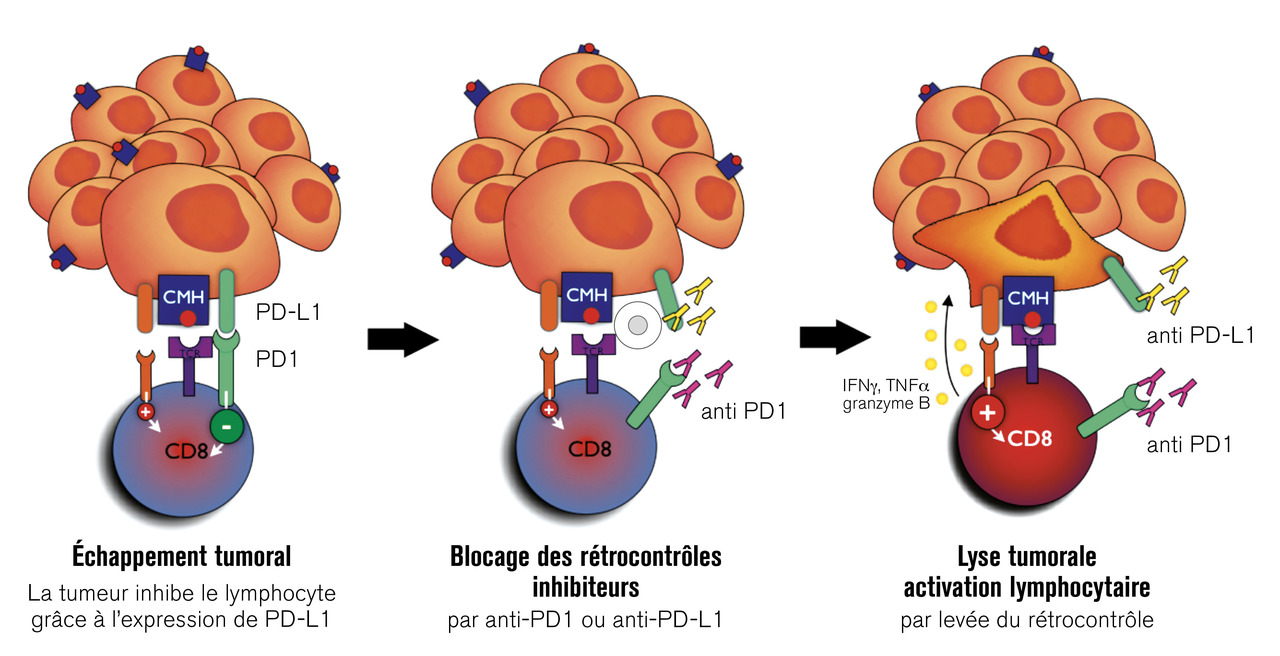
Parallèlement, un autre récepteur inhibiteur, PD-1, avait été identifié en 199212 et son caractère inhibiteur décrit en 1998 par l’équipe de Tasuku Honjo.13 PD-1 interagit avec ses ligands PD-L1 présents sur de nombreuses cellules et PD-L2. Comme CTLA-4, il n’est pas exprimé sur les lymphocytes T naïfs et apparaît après activation. Les souris PD-1 KO ont une lymphoprolifération marginale mais plutôt des manifestations auto-immunes. Honjo a montré que le blocage de la liaison PD-1-PD-L1 par des anticorps monoclonaux permettait d’augmenter la réponse immunitaire antitumorale et le traitement de tumeurs chez la souris (fig. 3).14
Cela a conduit à l’utilisation chez l’homme d’anticorps monoclonaux bloquant cette liaison PD-1-PD-L1 dans de nombreux cancers comme le mélanome, le cancer du poumon non à petites cellules, le cancer de la vessie, le cancer du rein, les cancers digestifs, les cancers de la sphère oto-rhino-laryngée, certains lymphomes...15 Parmi les dizaines d’anticorps en développement, certains ont déjà été reconnus par la FDA, deux anti-PD-1 (pembrolizumab et nivolumab) et un anti-PD-L1 (atézolimumab).
Même si les mécanismes d’action des anti-CTLA-4 et des anti-PD-1/PD-L1 font encore l’objet de nombreux travaux, il apparaît qu’ils sont différents. L’expression des molécules B7 sur les cellules présentatrices de l’antigène (cellules dendritiques) et non sur les cellules tumorales suggère que l’action des anti-CTLA-4 a lieu au niveau des organes lymphoïdes secondaires plus qu’au niveau des tumeurs. De plus, l’expression de CTLA-4 est plus importante au niveau des lymphocytes T CD4+ que des lymphocytes T CD8. Dans la mesure où CTLA-4 est fortement exprimé sur les lymphocytes T régulateurs, une déplétion de ces cellules au niveau des sites tumoraux pourrait rendre compte de l’efficacité des anticorps anti-CTLA-4. Cela semble vrai dans les modèles murins mais n’a pas été observé avec l’ipilimumab, ce qui pourrait justifier des modifications de son fragment Fc pour induire une cytotoxicité par antibody-dependant cellular cytotoxicity (ADCC). En raison de l’expression plus ubiquitaire des PD-L1, et en particulier au niveau des cellules tumorales, les anti-PD-1/PD-L1 semblent avoir une action plus périphérique, au niveau du micro-environnement tumoral, ce récepteur inhibiteur intervenant physiologiquement comme un inducteur de signaux « véto » anti-autoréactifs.
Aussi la combinaison de ces ICI apparaît synergique et justifie encore plus l’attribution conjointe du prix Nobel aux découvreurs de l’action anticancéreuse du blocage de ces deux récepteurs inhibiteurs. Des travaux récents ont d’ailleurs démontré que l’association de l’ipilimumab et du pembrolizumab était supérieure en termes de survie (58 % à 3 ans) à l’action de chacun des ICI séparément dans le traitement des mélanomes graves jusqu’alors peu sensibles aux thérapeutiques.15
Cette découverte a bien sûr suscité l’utilisation des anticorps anti-CTLA-4 chez l’homme. Un premier anticorps, l’ipilimumab, a été utilisé dans le traitement de cancers, et des résultats favorables dans 20 % des cas de mélanomes métastatiques ont été rapportés en 2010,11 justifiant sa reconnaissance comme premier inhibiteur du contrôle immunitaire (ICI), par la Food and Drug Administration (FDA) en 2011.Des travaux ultérieurs ont confirmé cette étude initiale, les résultats poolés dans le mélanome de stade III ou IV montrant chez 20 % des patients une réponse clinique durable avec un plateau débutant aux alentours de la troisième année, résultats bien supérieurs à ce qui était observé auparavant.
Parallèlement, un autre récepteur inhibiteur, PD-1, avait été identifié en 199212 et son caractère inhibiteur décrit en 1998 par l’équipe de Tasuku Honjo.13 PD-1 interagit avec ses ligands PD-L1 présents sur de nombreuses cellules et PD-L2. Comme CTLA-4, il n’est pas exprimé sur les lymphocytes T naïfs et apparaît après activation. Les souris PD-1 KO ont une lymphoprolifération marginale mais plutôt des manifestations auto-immunes. Honjo a montré que le blocage de la liaison PD-1-PD-L1 par des anticorps monoclonaux permettait d’augmenter la réponse immunitaire antitumorale et le traitement de tumeurs chez la souris (fig. 3).14
Cela a conduit à l’utilisation chez l’homme d’anticorps monoclonaux bloquant cette liaison PD-1-PD-L1 dans de nombreux cancers comme le mélanome, le cancer du poumon non à petites cellules, le cancer de la vessie, le cancer du rein, les cancers digestifs, les cancers de la sphère oto-rhino-laryngée, certains lymphomes...15 Parmi les dizaines d’anticorps en développement, certains ont déjà été reconnus par la FDA, deux anti-PD-1 (pembrolizumab et nivolumab) et un anti-PD-L1 (atézolimumab).
Même si les mécanismes d’action des anti-CTLA-4 et des anti-PD-1/PD-L1 font encore l’objet de nombreux travaux, il apparaît qu’ils sont différents. L’expression des molécules B7 sur les cellules présentatrices de l’antigène (cellules dendritiques) et non sur les cellules tumorales suggère que l’action des anti-CTLA-4 a lieu au niveau des organes lymphoïdes secondaires plus qu’au niveau des tumeurs. De plus, l’expression de CTLA-4 est plus importante au niveau des lymphocytes T CD4+ que des lymphocytes T CD8. Dans la mesure où CTLA-4 est fortement exprimé sur les lymphocytes T régulateurs, une déplétion de ces cellules au niveau des sites tumoraux pourrait rendre compte de l’efficacité des anticorps anti-CTLA-4. Cela semble vrai dans les modèles murins mais n’a pas été observé avec l’ipilimumab, ce qui pourrait justifier des modifications de son fragment Fc pour induire une cytotoxicité par antibody-dependant cellular cytotoxicity (ADCC). En raison de l’expression plus ubiquitaire des PD-L1, et en particulier au niveau des cellules tumorales, les anti-PD-1/PD-L1 semblent avoir une action plus périphérique, au niveau du micro-environnement tumoral, ce récepteur inhibiteur intervenant physiologiquement comme un inducteur de signaux « véto » anti-autoréactifs.
Aussi la combinaison de ces ICI apparaît synergique et justifie encore plus l’attribution conjointe du prix Nobel aux découvreurs de l’action anticancéreuse du blocage de ces deux récepteurs inhibiteurs. Des travaux récents ont d’ailleurs démontré que l’association de l’ipilimumab et du pembrolizumab était supérieure en termes de survie (58 % à 3 ans) à l’action de chacun des ICI séparément dans le traitement des mélanomes graves jusqu’alors peu sensibles aux thérapeutiques.15
Questions en suspens
Ainsi, les ICI ont apporté la preuve de leur efficacité clinique, mais bien des points restent à résoudre, en particulier la gestion des effets secondaires. En effet, en libérant les freins de la réponse immunitaire, les ICI peuvent entraîner des manifestations inflammatoires et auto-immunes, telles que thyroïdites, colites, hépatites…11, 15 Ces effets semblent plus fréquents avec les anti-CTLA-4 qu’avec les anti- PD-1/PD-L1 mais encore plus fréquents en cas d’association.
Par ailleurs, les ICI sont inefficaces chez un grand nombre de patients, en particulier des femmes. Il importe d’en comprendre les raisons et de définir des biomarqueurs d’efficacité. Une réponse immunitaire efficace nécessite la reconnaissance de « néoantigènes » tumoraux par des lymphocytes T spécifiques. Cela implique l’existence d’antigènes tumoraux présentés à des lymphocytes T capables de s’activer et de proliférer, puis de migrer et d’infiltrer la tumeur pour la détruire. L’absence d’une ou de plusieurs de ces conditions ainsi qu’un microenvironnement très immunosuppresseur pourrait rendre compte de l’absence d’efficacité des ICI.
Par ailleurs, d’autres récepteurs inhibiteurs ont été décrits : LAG-3 (lymphocyte activation gene 3), KIRs (killer inhibitory receptors), TIM-3 (T cell immunoglobulin and mucin 3), VISTA (V-domain-ig-containing suppressor of T-cell activation), TIGIT (T cell immunoglobulin and immunotyrosine inhibitory domain),16 dont le blocage ouvre autant de nouvelles pistes dans l’immunothérapie des cancers.17 Enfin, il est important de réfléchir à la prise en charge par la société du coût très élevé de ces traitements dont le marché est évalué en 2025 entre 45 et 100 milliards de dollars, même si on peut en imaginer la modulation en raison de la multiplication des anticorps thérapeutiques et de la compétition entre laboratoires pharmaceutiques.
Par ailleurs, les ICI sont inefficaces chez un grand nombre de patients, en particulier des femmes. Il importe d’en comprendre les raisons et de définir des biomarqueurs d’efficacité. Une réponse immunitaire efficace nécessite la reconnaissance de « néoantigènes » tumoraux par des lymphocytes T spécifiques. Cela implique l’existence d’antigènes tumoraux présentés à des lymphocytes T capables de s’activer et de proliférer, puis de migrer et d’infiltrer la tumeur pour la détruire. L’absence d’une ou de plusieurs de ces conditions ainsi qu’un microenvironnement très immunosuppresseur pourrait rendre compte de l’absence d’efficacité des ICI.
Par ailleurs, d’autres récepteurs inhibiteurs ont été décrits : LAG-3 (lymphocyte activation gene 3), KIRs (killer inhibitory receptors), TIM-3 (T cell immunoglobulin and mucin 3), VISTA (V-domain-ig-containing suppressor of T-cell activation), TIGIT (T cell immunoglobulin and immunotyrosine inhibitory domain),16 dont le blocage ouvre autant de nouvelles pistes dans l’immunothérapie des cancers.17 Enfin, il est important de réfléchir à la prise en charge par la société du coût très élevé de ces traitements dont le marché est évalué en 2025 entre 45 et 100 milliards de dollars, même si on peut en imaginer la modulation en raison de la multiplication des anticorps thérapeutiques et de la compétition entre laboratoires pharmaceutiques.
UNE RÉVOLUTION
Au total, la voie initiée par James Allison et Tasuku Honjo par le blocage des récepteurs inhibiteurs a conduit à une révolution médicale dans le traitement des cancers par l’immunothérapie, permettant aussi de nouvelles combinaisons thérapeutiques, avec non seulement les traitements habituels du cancer (chirurgie, chimiothérapie, radiothérapie) mais aussi avec les thérapies ciblées et celles visant à augmenter l’immunogénicité des tumeurs (par exemple par vaccination antitumorale par injection d’antigènes tumoraux ou viraux en cas de tumeurs viro-induites). V
Références
1. Mathé G, Amiel JL, Schwarzenberg L, et al. Adoptive immunotherapy of acute leukemia: experimental and clinical results. Cancer Research 1965;25:1525-31.
2. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996;86:973-83.
3. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004;10:909-15.
4. Dunn GP, Bruce AT, Ikeda H, Old LJ, and Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002;3:991-8.
5. Schwartz RH. A cell culture model for lymphocyte clonal anergy. Science 1990;248:1349-56.
6. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996;14:233-58.
7. Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activity. Immunity 1994;1:405-13.
8. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells costimulation. J Exp Med 1995;182:459-65.
9. Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction revealing a critical negative regulatory role of CTLA-4. Immunity 1995;5:541-7.
10. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734-6.
11. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23.
12. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11:3887-95.
13. Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol 1998;10:1563-72.
14. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol 2005;17:133-44.
15. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345-56.
16.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blocage: a common denominator approach to cancer therapy. Cancer Cell 2015;27:450-61.
17. Andre P, Denis C, Narmi-Mancinelli E, et al. Anti-NKG2A mab is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018;175:1-13.
2. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996;86:973-83.
3. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004;10:909-15.
4. Dunn GP, Bruce AT, Ikeda H, Old LJ, and Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002;3:991-8.
5. Schwartz RH. A cell culture model for lymphocyte clonal anergy. Science 1990;248:1349-56.
6. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996;14:233-58.
7. Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activity. Immunity 1994;1:405-13.
8. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells costimulation. J Exp Med 1995;182:459-65.
9. Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction revealing a critical negative regulatory role of CTLA-4. Immunity 1995;5:541-7.
10. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734-6.
11. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23.
12. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11:3887-95.
13. Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol 1998;10:1563-72.
14. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol 2005;17:133-44.
15. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345-56.
16.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blocage: a common denominator approach to cancer therapy. Cancer Cell 2015;27:450-61.
17. Andre P, Denis C, Narmi-Mancinelli E, et al. Anti-NKG2A mab is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018;175:1-13.