Il touche avec prédilection la femme en période d’activité génitale (sex-ratio de 9 femmes pour 1 homme). Il est habituellement plus sévère chez l’homme et dans la population antillaise.
Pathogénie : complexe
La cascade pathogène débuterait par une réaction inadaptée des cellules dendritiques (qui présentent les antigènes au système immunitaire) en présence de résidus moléculaires de l’apoptose normale des cellules. Cela aboutirait à l’activation de lymphocytes B et C autoréactifs et à l’accumulation de complexes immuns (auto-anticorps et protéines cellulaires) dans les organes. Les anticorps antiphospholipides, particulièrement néfastes, s’attaquent aux vaisseaux (cellules endothéliales), entraînant une hypercoagulabilité responsable de thromboses.
Une prédisposition génétique existe, mais les mutations mono- géniques des protéines C2 ou C4 du complément par exemple, sont exceptionnelles. Les atteintes polygéniques constituent la majorité des observations chez l’adulte.
Des éléments extérieurs favorisent un LES ou ses poussées : exposition solaire, médicaments, stress, infection virale (EBV), hormones sexuelles (grossesse, prise d’estrogènes). Un terrain génétique favorable est considéré comme nécessaire pour qu’ils puissent déclencher la maladie.
Certains médicaments (
Clinique : polymorphe
Manifestations cutanées
Trois types de lésions spécifiques sont décrits. Le lupus aigu est caractérisé par le vespertilio de topographie symétrique ou en ailes de papillon (
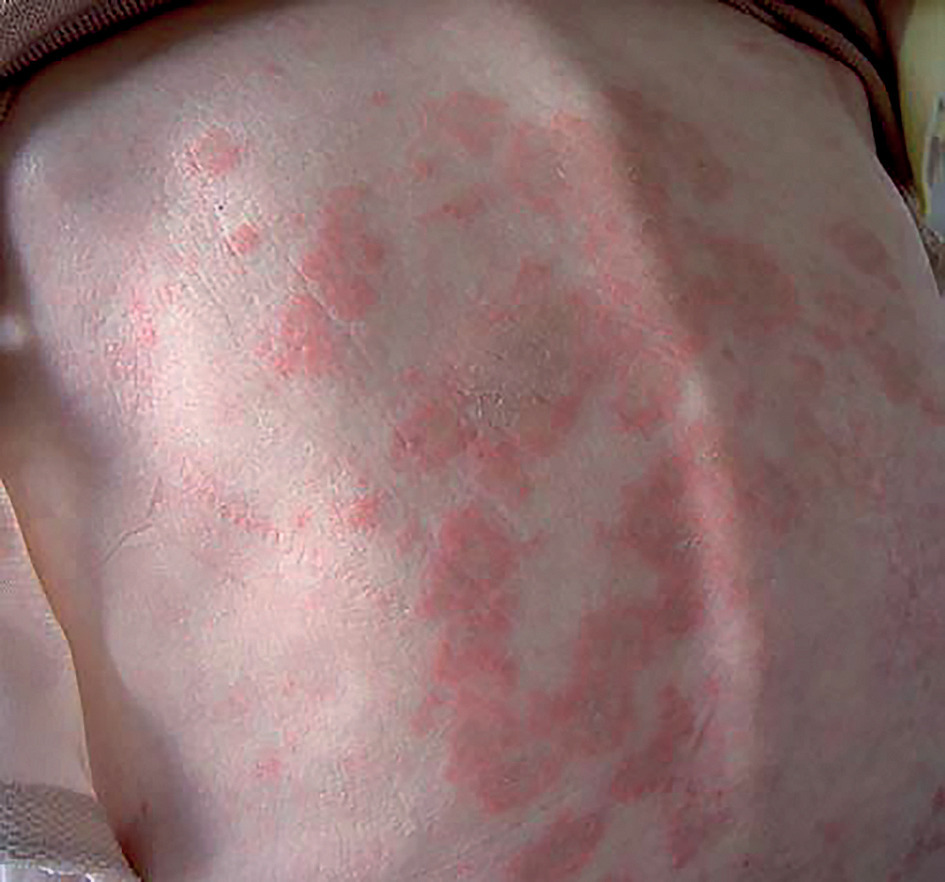
Le lupus chronique, le plus souvent discoïde, se manifeste par des plaques érythémateuses, des squames et une atrophie cicatricielle. Il siège au visage, aux oreilles, aux mains.
Livedo, phénomène de Raynaud, urticaire, purpura, ulcère de jambes sont parfois associés.
Manifestations articulaires
Manifestations neurologiques
Le diagnostic est difficile dans ces derniers cas quand l’IRM cérébrale est normale. Les manifestations directement liées au lupus doivent être distinguées des complications de la corticothérapie ou d’une dépression liée à une maladie chronique. Une atteinte démyélinisante et neuropsychiatrique est considérée comme grave et péjorative.
Manifestations rénales
La principale atteinte est glomérulaire, mais ce n’est pas la seule : il peut s’agir d’une néphropathie vasculaire (souvent associée à un SAPL) ou interstitielle.
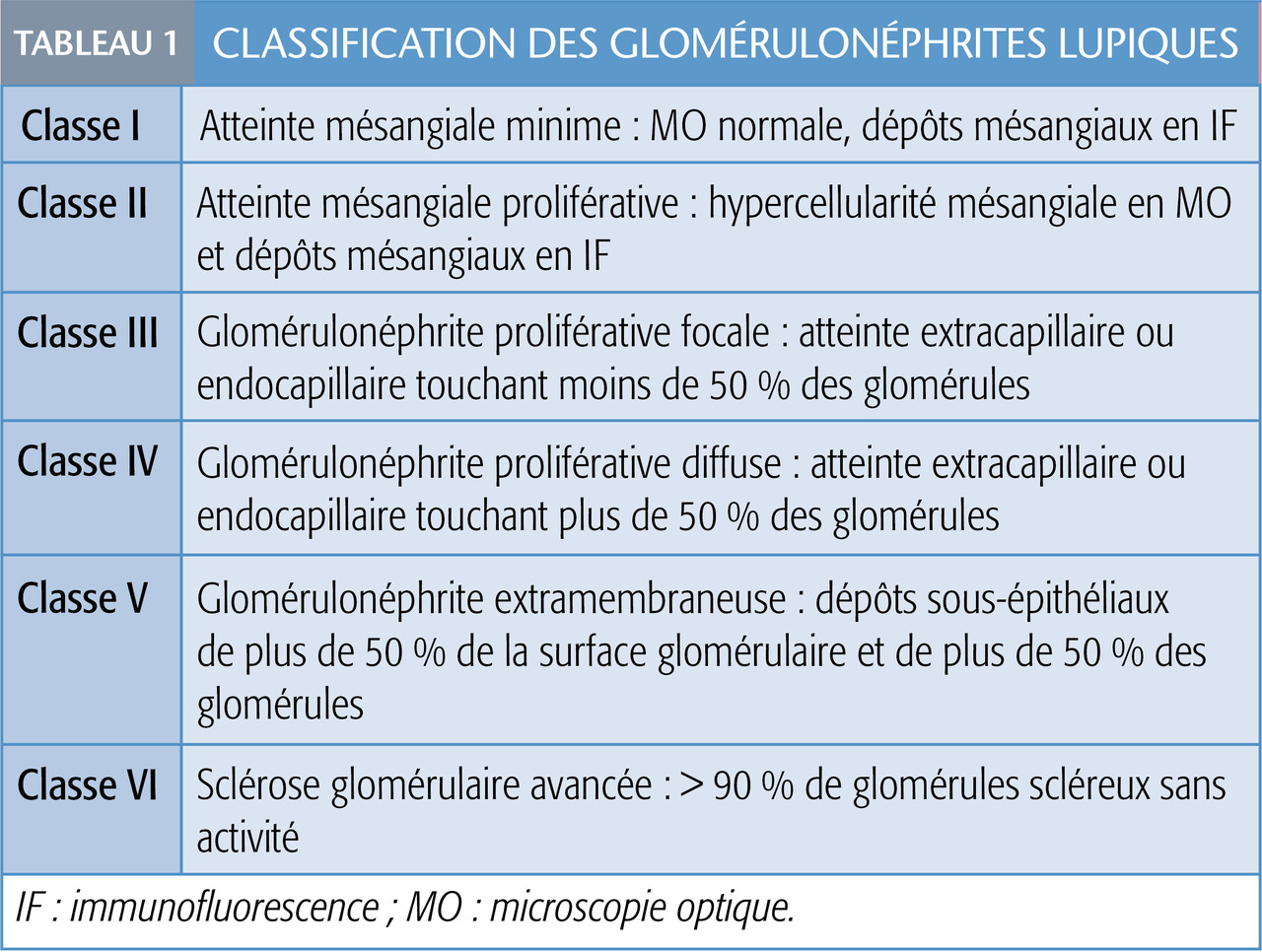
L’histologie est cruciale (
Manifestations cardiaques
Manifestations pulmonaires
Pneumopathie interstitielle ou hypertension artérielle pulmonaire sont plus rares.
Autres éléments
L’analyse histologique d’adénopathies périphériques inexpliquées met en évidence une inflammation non spécifique : infiltrat lymphocytaire non tumoral, hyperplasie lymphocytaire réactionnelle ou aspect de maladie de Kikuchi.
Syndromes auto-immuns associés
Fréquent, le syndrome des antiphospholipides peut se manifester par : thromboses veineuses et/ou artérielles, complications obstétricales (fausses couches récurrentes précoces, morts fœtales in utero, prééclampsie ou prématurité en rapport avec une insuffisance placentaire). Il est caractérisé par des anticorps antiphospholipides persistant à au moins 12 semaines.
Parmi les atteintes d’organes, l’association à une thyroïdite auto-immune est l’une des plus courantes.
Bilan biologique
Anomalies non spécifiques
Il n’y a pas habituellement de syndrome inflammatoire au cours des poussées, sauf lors d’une atteinte séreuse ou de survenue d’une infection.
L’hémogramme peut révéler des cytopénies, le plus souvent auto-immunes (anémie hémolytique, thrombopénie immunologique, neutropénie auto-immune), mais d’autres causes sont possibles (carence en fer, micro-angiopathie thrombotique, syndrome d’activation macrophagique). Une lymphopénie est fréquente.
Un TCA allongé peut être dû à un anticoagulant circulant lupique.
Stigmates immunologiques
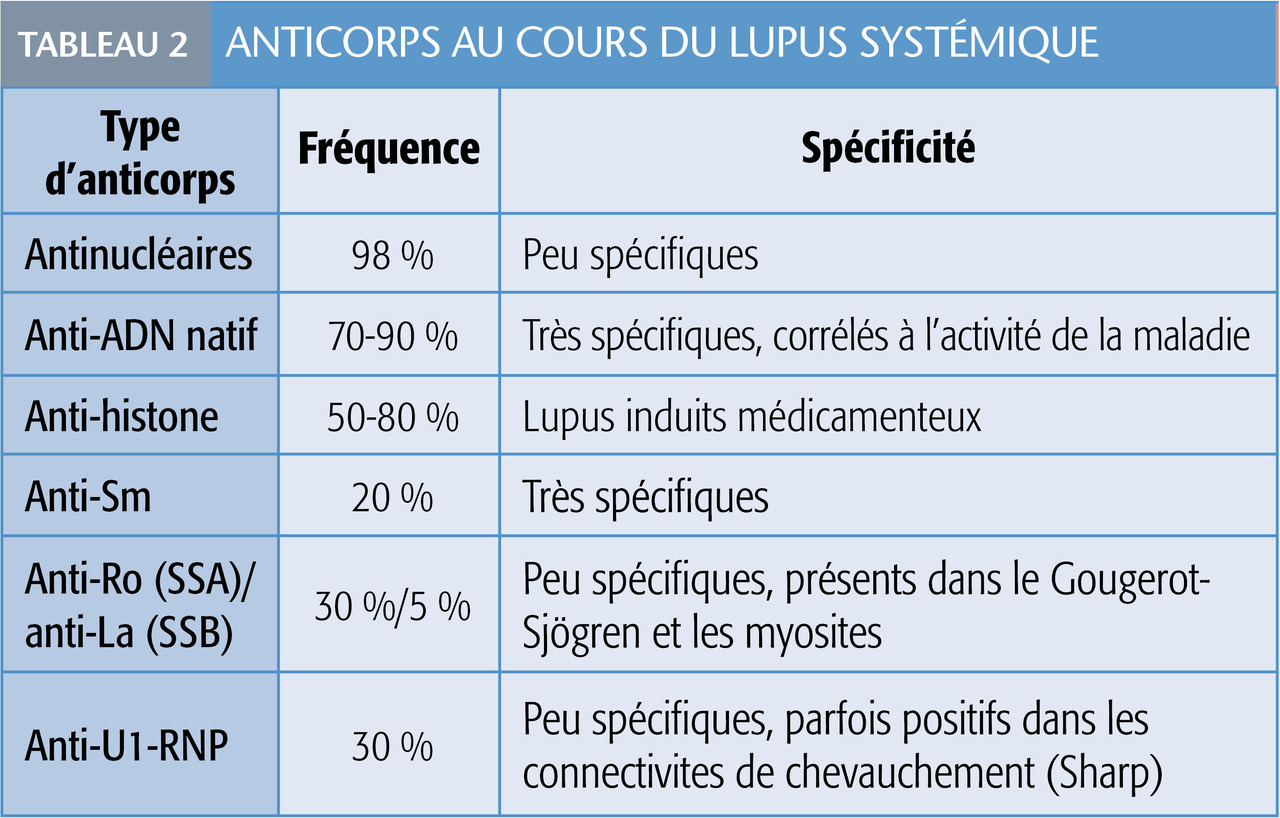
Les anticorps anti-ADN natifs reflètent l’activité de la maladie ; on les recherche par test ELISA ou test de Farr.
D’autres anticorps peuvent être présents, en particulier anti- thyroïdiens et antiphospholipides.
Lors des poussées lupiques, l’activation du complément se fait par la voie classique avec formation de complexes immuns ou via une cryoglobulinémie associée, avec baisse du CH50, C3 ou C4 très évocatrice. Une activation permanente fait suspecter un déficit génétique des protéines du complément (rare).
Diagnostic : difficile
Cette hétérogénéité rend le diagnostic difficile, surtout quand les signes sont peu spécifiques (arthralgies, atteintes séreuses ou neurologiques).
Certains doivent alerter : lésions cutanées évocatrices, arthralgies moins spécifiques imposant un bilan immunologique systématique chez une femme jeune.
Parmi les 11 critères définis par l’ACR, 4 suffisent à retenir le diagnostic (
Évolution – pronostic
La pathologie est habituellement plus sévère chez l’enfant, les hommes et les sujets à peau noire.
Les glomérulonéphrites (stades III et IV) et les atteintes du système nerveux central sont les formes les plus graves et requièrent une immunosuppression.
L’éducation et l’information du patient, une bonne observance (hydroxychloroquine), l’éviction des facteurs favorisant les poussées sont essentiels à l’amélioration du pronostic.
Les complications des traitements sont à anticiper autant que faire se peut : rétinopathie sous hydroxychloroquine, infections et ostéoporose favorisées par les corticoïdes et les immuno- suppresseurs, risque néoplasique (hémopathies) induit.
Prise en charge
En traitement d’attaque (lupus actif) et en prévention des poussées, l’immunomodulation comporte une corticothérapie à doses variables selon l’atteinte, avec ou sans immunosuppresseur.
L’hydroxychloroquine (Plaquenil) est indispensable en thérapie de fond quelles que soient la forme et l’activité de la maladie, avec une surveillance ophtalmologique (commune aux antipaludéens de synthèse) et l’arrêt du tabac (qui diminue son efficacité).
En seconde ligne peuvent être proposés le méthotrexate contre l’atteinte articulaire et le thalidomide dans les formes cutanées.
Dans les troubles modérés cutanéo-articulaires, le bélimumab (Benlysta, anti-BAFF monoclonal) a récemment obtenu l’AMM en injection mensuelle intraveineuse. Il a une bonne efficacité en cas d’échec des traitements précédents, en particulier pour les atteintes articulaires et cutanées ; la prescription est pour le moment réservée aux spécialistes.
Les formes sévères justifient corticothérapie (Cortancyl) et immunosuppression d’emblée : notamment les glomérulonéphrites lupiques stades III et IV et les atteintes neurologiques centrales. Au cas pas cas et selon l’âge (surtout chez la femme), l’immunosuppresseur sera le mycophénolate mofétil (2 à 3 g/j) ou le cyclophosphamide intraveineux, et ce pendant 3 à 5 ans (durée non codifiée).
Un syndrome des antiphospholipides avec thrombose artérielle ou veineuse implique une anticoagulation définitive à dose curative.
Mesures préventives
La grossesse est autorisée si le lupus est quiescent depuis au moins 6 mois, avec un bilan initial comportant : complément, anticorps anti-ADN et antiphospholipides (aspirine en l’absence de complications obstétricales ou thrombotiques antérieures) et anticorps anti-SSA/SSB (si positifs : risque de lupus néonatal à surveiller par échographie cardiaque fœtale pour dépister bloc auriculo-ventriculaire et cardiomyopathie).
La maîtrise des risques liés au traitement est nécessaire : ostéoporose cortisonique (supplémentation vitamino-calcique et anti-ostéoporotique selon les recommandations), infections liées aux immunosuppresseurs (prévention antipneumocystose et herpès virus), atteinte rétinienne sous hydroxychloroquine, imposant un bilan ophtalmologique annuel (fond d’œil, champ visuel central et électrorétinogramme multifocal).
L’éducation et l’information portent sur les signes d’une poussée, l’observance (vérifiée par l’hydroxychloroquinémie), les facteurs déclenchants/favorisants : photoprotection +++, éviction des médicaments inducteurs, sevrage tabagique, contraception adaptée. Les estroprogestatifs sont contre-indiqués et on peut recourir aux microprogestatifs, aux moyens mécaniques ou au stérilet en l’absence d’immunosuppression sévère.
Enfin, la déclaration d’affection de longue durée (ALD 21) est indispensable.
1. Molécules responsables de lupus érythémateux induits
• Isoniazide
• Phénothiazines
• Quinidine• Minocycline
• Interféron alpha•
Antagonistes du TNFa
• Hydralazine
• D-pénicillamine
• Sulfasalazine
2. Critères du lupus érythémateux systémique (ACR* 1997)
✔ Éruption cutanée malaire (vespertilio)
✔ Lupus discoïde
✔ Photosensibilité
✔ Ulcérations orales ou nasopharyngées
✔ Arthrite non érosive périphérique
✔ Pleurésie ou péricardite
✔ Protéinurie de plus de 0,5 g/24 heures
✔ Convulsions ou psychose
✔ Anémie auto-immune et/ou leucopénie < 4 000/mm3 et/ou lymphopénie < 1 500 /mm3 et/ou thrombopénie < 100 000/mm3
✔ Anticorps anti-ADN natifs et/ou anti-Sm et/ou anticoagulant circulant (ACC) ou anticorps anticardiolipine (IgG ou IgM) et/ou sérologie syphilitique dissociée ✔ Anticorps antinucléaires en l’absence de médicaments inducteurs
*American College of Rheumatology
Perspectives thérapeutiques
Le traitement fait appel le plus souvent à l’hydroxychloroquine et à la corticothérapie, ainsi qu’aux immunosuppresseurs de type cyclophosphamide et mycophénolate mofétil.
Le développement des biothérapies a ouvert de nouvelles pistes : le rituximab, anticorps anti-CD20 antinéoplasique indiqué notamment dans la polyarthrite rhumatoïde, bien que n’ayant pas l’AMM dans le lupus, peut être efficace dans certaines formes de la maladie ; le benlysta, anticorps dirigé contre la cytokine BAFF (B-cell activating factor), est commercialisé depuis 2012 ; enfin, des molécules anti-interférons sont en développement avec des résultats prometteurs.
– Mathian A, Arnaud L, Amoura Z. Physiopathologie du lupus érythémateux systémique : le point en 2014. Rev Med Interne 2014;35:503-11.
– Mathian A, Arnaud L, Amoura Z. Prise en charge du lupus systémique. Rev Prat 2011;61:1263-70.
– Hello M, Senand R, Hamidou M. Lupus érythémateux systémique. Rev Prat Med Gen 2011;25:595-601.
Dans cet article
 Encadrés
Encadrés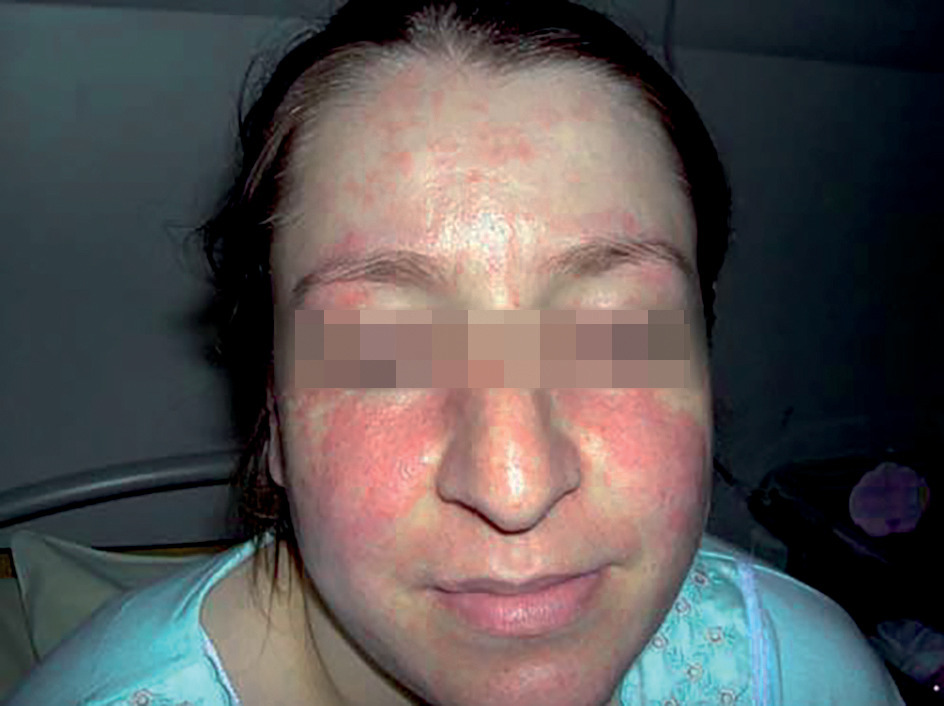
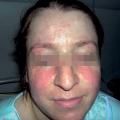
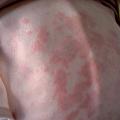


Le lupus est le plus souvent une pathologie bénigne se manifestant par des douleurs articulaires et des lésions cutanées typiques.
Des anticorps antinucléaires à un taux significatif sont un élément d’orientation non spécifique.
Les anti-ADN, moins sensibles mais plus spécifiques, reflètent l’activité de la maladie.
L’atteinte rénale peut être grave et impose une surveillance régulière de la protéinurie.
L’hydroxychloroquine, indispensable, diminue le risque de récidive et doit être prescrite (et surveillée) au long cours.
L’éviction/gestion des déclencheurs des poussées (exposition solaire, stress, tabac, contraception, grossesse) est capitale.