Dépister (notamment chez l’homme jeune) et surveiller, en collaboration avec le néphrologue.
La maladie de Berger, ou néphropathie à dépôts mésangiaux d’IgA (NIgA), est la glomérulonéphrite primitive la plus fréquente.
Si l’incidence de cette pathologie est relativement faible, estimée à 2,5/100 000 habitants en France,1 la prévalence est élevée, de l’ordre de 1 % de la population. Cette différence est liée au fait qu’elle débute chez l’adulte jeune et évolue de façon chronique durant toute la vie. Les disparités géographiques sont extrêmement marquées, avec un gradient Est-Ouest et Nord-Sud : la fréquence est importante en Asie (5,7/100 000 au Japon) et très faible en Afrique (0,2/100 000).
Si l’incidence de cette pathologie est relativement faible, estimée à 2,5/100 000 habitants en France,1 la prévalence est élevée, de l’ordre de 1 % de la population. Cette différence est liée au fait qu’elle débute chez l’adulte jeune et évolue de façon chronique durant toute la vie. Les disparités géographiques sont extrêmement marquées, avec un gradient Est-Ouest et Nord-Sud : la fréquence est importante en Asie (5,7/100 000 au Japon) et très faible en Afrique (0,2/100 000).
Physiopathologie
Elle n’est pas complètement connue. Une prédisposition génétique est impliquée (maladie polygénique),2 mais elle est insuffisante à elle seule. S’y associent de probables facteurs environnementaux déterminant des anomalies de production et de clairance des IgA1 dans le secteur plasmatique. Ces dernières sont quantitatives (augmentation de leur taux) et qualitatives (excès de formes anormalement O-glycosylées [IgA1-dg] au niveau de leur région charnière). Ces IgA1 anormales se lient à des IgG ou IgA anti-IgA1-dg et/ou à la portion extracellulaire clivée du récepteur aux IgA,3, 4 formant des complexes immuns de haut poids moléculaire qui se déposent au niveau de la partie mésangiale des glomérules. Cela induit une réponse mettant en jeu le complément, la sécrétion de cytokines par les cellules mésangiales et la migration de monocytes/macrophages. Cette réaction inflammatoire évolue vers des lésions scléreuses glomérulaires touchant l’ensemble du parenchyme rénal.
Un diagnostic histologique
La manifestation la plus classique est la survenue d’hématuries macroscopiques pendant des épisodes d’infections muqueuses (ORL en particulier). Brunes, parfois rouges, sans aucun caillot, elles concernent la totalité de la miction, apparaissent dans les jours suivant le début de l’infection et perdurent jusqu’à sa disparition. Elles se distinguent ainsi des hématuries associées à une glomérulonéphrite aiguë post-streptococcique, qui surviennent au moins 10 jours après l’infection. La récidive de tels épisodes est très évocatrice. Rarement, une insuffisance rénale aiguë régressive complique le tableau, liée à une obstruction tubulaire par les cylindres hématiques.
Le plus souvent, aucun symptôme n’est rapporté par le patient, et c’est à l’occasion d’un bilan qu’on fait le diagnostic. Par exemple, une microhématurie et/ou une protéinurie peut être mise en évidence à la bandelette urinaire au décours d’une visite de médecine du travail. La circonstance révélatrice peut aussi être une analyse systématique des urines, une évaluation de la fonction rénale (estimation du DFG par la créatinine) ou un bilan d’HTA prescrits par le généraliste.
L’exploration minimale comporte une protéinurie des 24 heures ou rapportée à la créatininurie sur échantillon, un examen cytobactériologique des urines, la prise de la PA et l’estimation du DFG (créatinine).
Une biopsie rénale est réalisée devant l’association protéinurie + hématurie ou DFGe < 60 mL/min/1,73 m2 + hématurie. En cas de microhématurie isolée (> 10 éléments/mm3), elle n’est pas nécessaire, mais un suivi spécialisé est recommandé. Une hématurie macroscopique récidivante avec sédiment urinaire et fonctionnement rénal normaux entre les épisodes est une indication plus discutée, le diagnostic étant alors presque certain, et le pronostic plutôt bon.
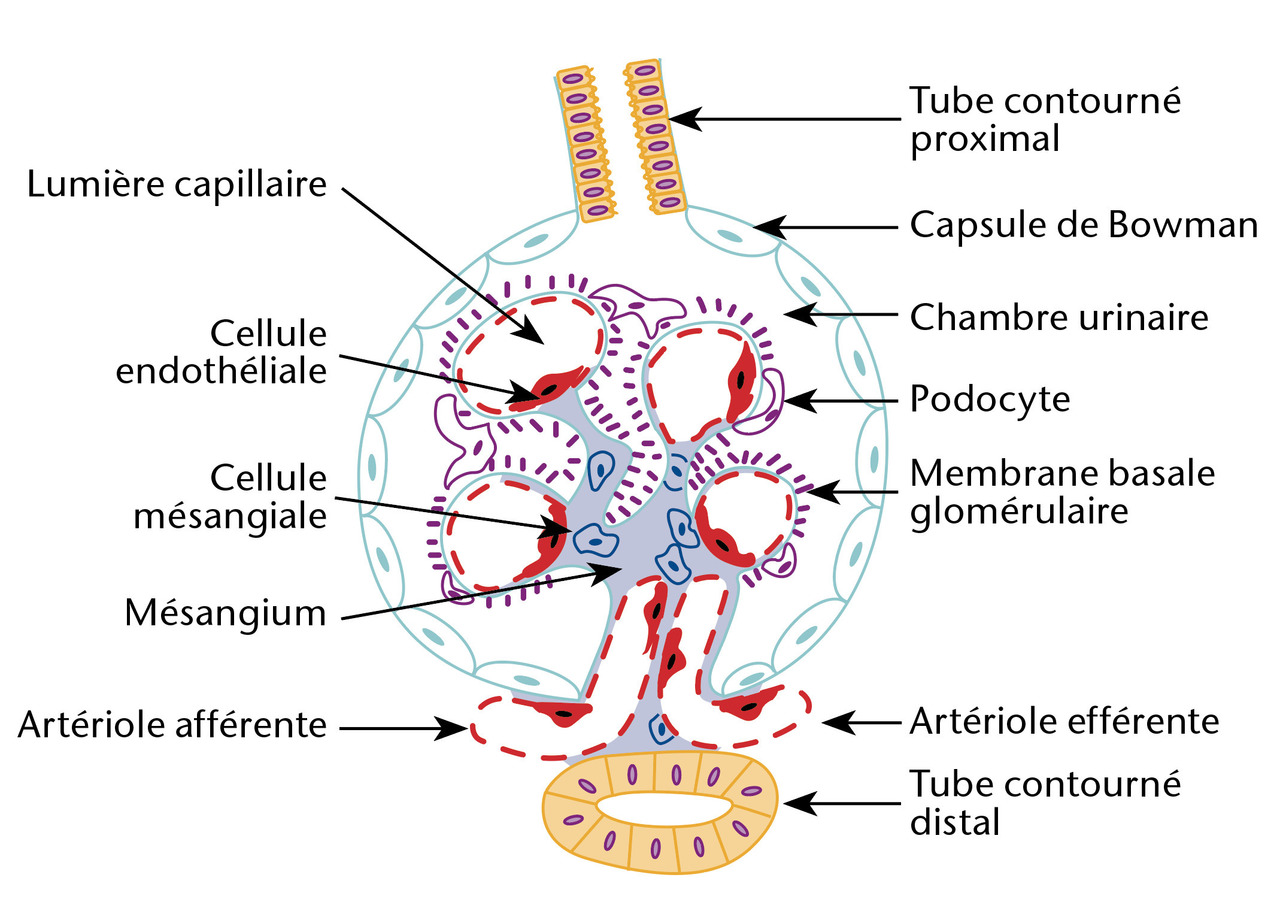
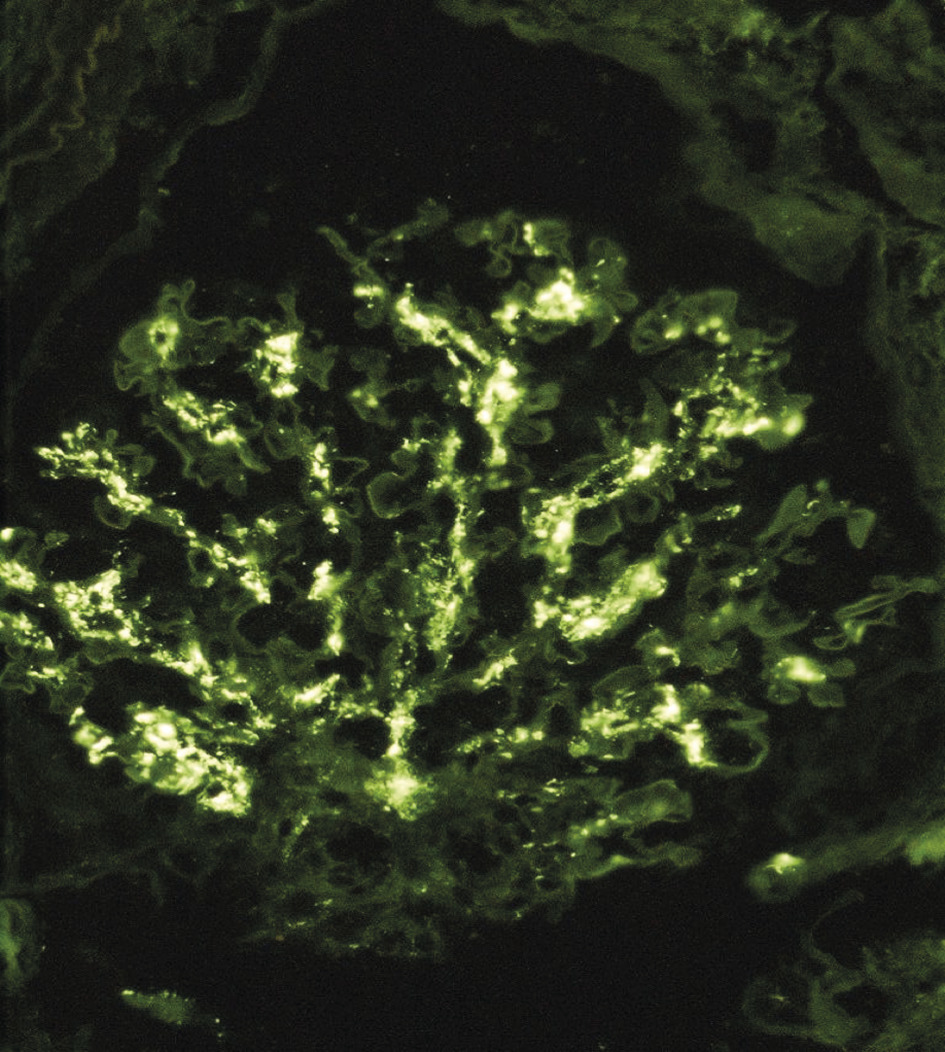
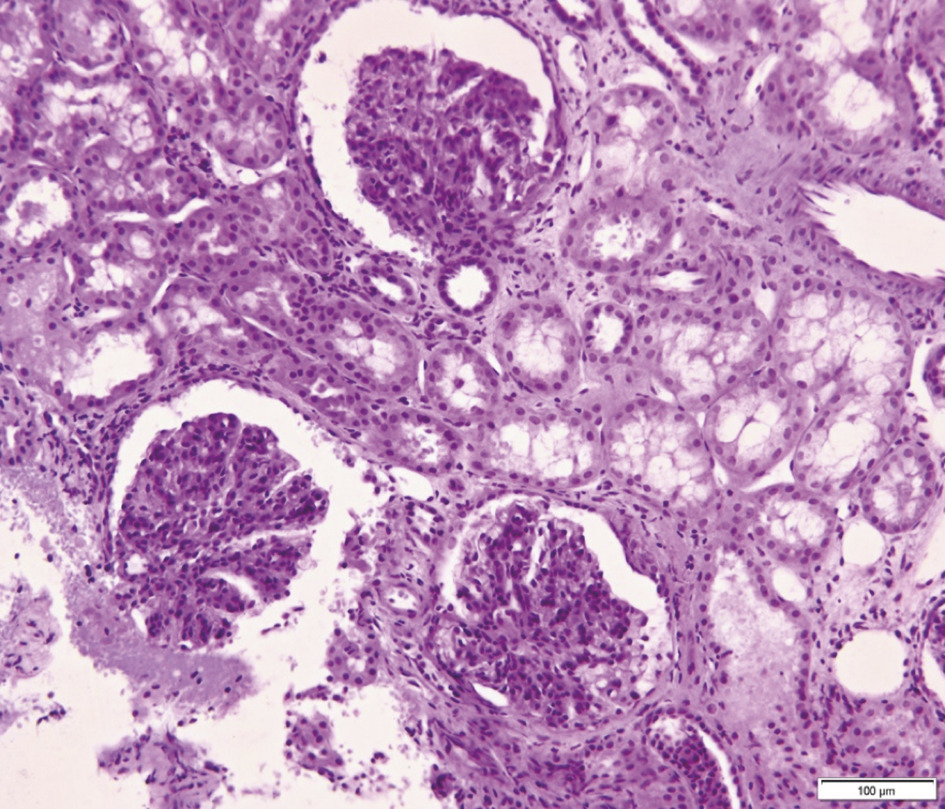
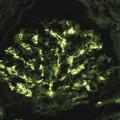
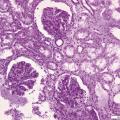
L’immunomarquage met en évidence des dépôts d’IgA au sein du mésangium, structure située entre les capillaires glomérulaires (fig. 2), diffus et volontiers associés à des agrégats de C3. L’analyse morphologique conventionnelle montre des lésions glomérulaires inconstantes, le plus souvent segmentaires (touchant moins de 50 % du glomérule) et focales (< 50 % des glomérules) : épaississement du mésangium, prolifération mésangiale (fig. 3), endocapillaire ou extracapillaire, hyalinose segmentaire et focale. Les glomérules peuvent devenir complètement scléreux, prenant l’aspect de « pains à cacheter ». Des lésions vasculaires et tubulo-interstitielles sont possibles. Enfin, on retrouve fréquemment des hématies dans la chambre urinaire glomérulaire ou des cylindres hématiques dans les lumières des tubes. Ces lésions morphologiques ne sont pas utiles au diagnostic, mais certaines ont un impact pronostique bien établi.
Le plus souvent, aucun symptôme n’est rapporté par le patient, et c’est à l’occasion d’un bilan qu’on fait le diagnostic. Par exemple, une microhématurie et/ou une protéinurie peut être mise en évidence à la bandelette urinaire au décours d’une visite de médecine du travail. La circonstance révélatrice peut aussi être une analyse systématique des urines, une évaluation de la fonction rénale (estimation du DFG par la créatinine) ou un bilan d’HTA prescrits par le généraliste.
L’exploration minimale comporte une protéinurie des 24 heures ou rapportée à la créatininurie sur échantillon, un examen cytobactériologique des urines, la prise de la PA et l’estimation du DFG (créatinine).
Une biopsie rénale est réalisée devant l’association protéinurie + hématurie ou DFGe < 60 mL/min/1,73 m2 + hématurie. En cas de microhématurie isolée (> 10 éléments/mm3), elle n’est pas nécessaire, mais un suivi spécialisé est recommandé. Une hématurie macroscopique récidivante avec sédiment urinaire et fonctionnement rénal normaux entre les épisodes est une indication plus discutée, le diagnostic étant alors presque certain, et le pronostic plutôt bon.
L’immunomarquage met en évidence des dépôts d’IgA au sein du mésangium, structure située entre les capillaires glomérulaires (fig. 2), diffus et volontiers associés à des agrégats de C3. L’analyse morphologique conventionnelle montre des lésions glomérulaires inconstantes, le plus souvent segmentaires (touchant moins de 50 % du glomérule) et focales (< 50 % des glomérules) : épaississement du mésangium, prolifération mésangiale (fig. 3), endocapillaire ou extracapillaire, hyalinose segmentaire et focale. Les glomérules peuvent devenir complètement scléreux, prenant l’aspect de « pains à cacheter ». Des lésions vasculaires et tubulo-interstitielles sont possibles. Enfin, on retrouve fréquemment des hématies dans la chambre urinaire glomérulaire ou des cylindres hématiques dans les lumières des tubes. Ces lésions morphologiques ne sont pas utiles au diagnostic, mais certaines ont un impact pronostique bien établi.
évolution : hétérogène
Dans 30 % des cas, cette maladie évolue vers l’insuffisance rénale chronique de stade 5 ou terminale (< 15 mL/min/1,73 m2), 20 ans après le diagnostic, mais de fortes disparités existent.
Les formes à microhématurie isolée ont un pronostic excellent, contrairement à celles associant forte protéinurie, HTA, altération du DFGe et lésions histologiques sévères. Devant cette grande hétérogénéité, une démarche de stratification du risque est indispensable afin d’adapter la prise en charge.
Selon de nombreuses études : HTA, protéinurie > 1 g/24 h, DFGe < 60 mL/min/1,73 m2 sont des facteurs de risque indépendants les uns des autres.
Certains marqueurs histologiques ont également un impact pronostique. Selon la classification d’Oxford, les lésions élémentaires associées à une évolution vers le stade 5 et le doublement de la créatinine5 sont : prolifération mésangiale (notée M : 0 ou 1), endocapillaire (E : 0 ou 1), lésion de hyalinose segmentaire focale (S : 0 ou 1), atrophie tubulaire/fibrose interstitielle (T : 0, 1 ou 2), prolifération extracapillaire (C : 0, 1 ou 2). Chacune de ces atteintes est associée à un risque évolutif péjoratif, indépendamment des éléments cliniques.
Selon le score développé au CHU de Saint-étienne, un risque rénal absolu (RRA) peut être calculé, variant de 0 à 3 selon la présence d’une HTA, d’une protéinurie > 1 g/24 h et de lésions histologiques (chacun de ces facteurs = 1 point).6 Le risque d’évolution vers une IR de stade 5 ou le décès est de 4 % à 20 ans si le RRA est nul, alors qu’il est de 64 % s’il est estimé à 3.
En cas de progression vers une IR terminale, la transplantation rénale est fréquente, car les patients concernés sont jeunes (environ 50 ans) et ont peu de comorbidités. Cependant, des dépôts mésangiaux d’IgA sont retrouvés dans environ 25 % des biopsies systématiques du greffon à 1 an, avec 25 % de rechutes clinico-histologiques à 5 ans (c’est-à-dire s’associant à une protéinurie et une hématurie par exemple). Cette récidive est considérée comme bénigne, mais elle est tout de même responsable de pertes de greffons, notamment 10 ans après la transplantation.Le jeune âge de début de la maladie est associé à un risque plus élevé de récidive sur le transplant. La persistance d’une cortico- thérapie à 1 an de la transplantation et le recours à une induction immunosuppressive par serum antilymphocytaire est en revanche associé à un risque plus faible de récidive.
Les formes à microhématurie isolée ont un pronostic excellent, contrairement à celles associant forte protéinurie, HTA, altération du DFGe et lésions histologiques sévères. Devant cette grande hétérogénéité, une démarche de stratification du risque est indispensable afin d’adapter la prise en charge.
Selon de nombreuses études : HTA, protéinurie > 1 g/24 h, DFGe < 60 mL/min/1,73 m2 sont des facteurs de risque indépendants les uns des autres.
Certains marqueurs histologiques ont également un impact pronostique. Selon la classification d’Oxford, les lésions élémentaires associées à une évolution vers le stade 5 et le doublement de la créatinine5 sont : prolifération mésangiale (notée M : 0 ou 1), endocapillaire (E : 0 ou 1), lésion de hyalinose segmentaire focale (S : 0 ou 1), atrophie tubulaire/fibrose interstitielle (T : 0, 1 ou 2), prolifération extracapillaire (C : 0, 1 ou 2). Chacune de ces atteintes est associée à un risque évolutif péjoratif, indépendamment des éléments cliniques.
Selon le score développé au CHU de Saint-étienne, un risque rénal absolu (RRA) peut être calculé, variant de 0 à 3 selon la présence d’une HTA, d’une protéinurie > 1 g/24 h et de lésions histologiques (chacun de ces facteurs = 1 point).6 Le risque d’évolution vers une IR de stade 5 ou le décès est de 4 % à 20 ans si le RRA est nul, alors qu’il est de 64 % s’il est estimé à 3.
En cas de progression vers une IR terminale, la transplantation rénale est fréquente, car les patients concernés sont jeunes (environ 50 ans) et ont peu de comorbidités. Cependant, des dépôts mésangiaux d’IgA sont retrouvés dans environ 25 % des biopsies systématiques du greffon à 1 an, avec 25 % de rechutes clinico-histologiques à 5 ans (c’est-à-dire s’associant à une protéinurie et une hématurie par exemple). Cette récidive est considérée comme bénigne, mais elle est tout de même responsable de pertes de greffons, notamment 10 ans après la transplantation.
Prise en charge7
Si le risque d’évolution vers l’IR terminale est faible (microhématurie isolée, protéinurie < 0,5 g/j, absence d’HTA et DFG > 60 mL/min/1,73 m2), de simples mesures hygiénodiététiques et un suivi spécialisé espacé (tous les 2 ans) sont suffisants.
En cas de risque intermédiaire ou élevé (protéinurie > 0,5 g/j au diagnostic, plus ou moins associée à une HTA et une altération du DFGe), des mesures générales de néphroprotection sont recommandées. Une PA < 130/80 mmHg doit être obtenue avec des moyens non pharmacologiques (apports sodés limités à 6 g/j, activité physique, correction du surpoids, arrêt d’une intoxication éthylique chronique) et des thérapies médicamenteuses.
Les bloqueurs du système rénine-angiotensine sont indiqués – au premier rang desquels les IEC. Toutefois, un antagoniste des récepteurs de type 1 de l’angiotensine II (ARA II) peut être également prescrit (recommandation de niveau élevé lorsque la protéinurie initiale est > 1 g/24 h et suggéré si elle est située entre 0,5 et 1 g/24 h). L’association de ces 2 familles (double blocage) est possible, mais elle doit être initiée et surveillée par le néphrologue (risque accru d’instabilité de la fonction rénale).
Une restriction protidique modérée (0,6-0,8 g/kg/24 h) est préconisée, même si le niveau de preuve de cette recommandation est modeste.
Dans les formes les plus sévères, à risque évolutif élevé, des immunosuppresseurs sont envisageables. La corticothérapie a fait l’objet de plusieurs études contrôlées/randomisées et de méta-analyses, qui montrent un bénéfice en termes de réduction de la protéinurie et d’évolution de la fonction rénale ; cependant, dans ces essais les patients n’ont pas un contrôle tensionnel suffisant, et une faible fraction est traitée par IEC ou ARA II. Deux travaux récents ont remis en cause la place des corticoïdes au cours de la NIgA. Dans STOP-IGAN (étude randomisée contrôlée avec un schéma complexe vs placebo), une corticothérapie (si DFG > 60 mL/min/1,73 m2) ou le cyclophosphamide relayé par azathioprine (si DFG compris entre 30 et 60 mL/min/1,73 m2) était instauré lorsque la protéinurie restait > 0,75 g/j après 6 mois à 3 ans de néphroprotection maximale.8 Le critère de jugement principal a été atteint (rémission de la protéinurie, stabilité de la fonction rénale), mais aucun bénéfice n’a été retrouvé en termes d’évolution du DFG à 3 ans. Les auteurs concluent à l’absence d’effets significatifs au long cours sur la fonction rénale. Néanmoins, cette étude a de nombreux biais, notamment le fait que les patients pouvaient être inclus de nombreuses années après le diagnostic, sélectionnant des malades très stables. L’essai TESTING montre l’efficacité de la corticothérapie : régression de la protéinurie et gain de fonction rénale, versus placebo ;9 cependant, il a été interrompu précocement en raison d’un surcroît d’effets secondaires graves, essentiellement des infections, chez les patients traités.
Ainsi, les recommandations KDIGO ne proposent la cortico- thérapie (niveau de suggestion, 2C) qu’en cas de protéinurie persistante > 1 g/j malgré 3 à 6 mois de traitement antihypertenseur et antiprotéinurique (s’appuyant sur les IEC/ARA II si le DFG est > 50 mL/min/1,73 m2). Son utilisation est donc très débattue, mais elle n’est pas abandonnée. Elle est réservée à des patients jeunes, ayant une protéinurie > 1 g/j malgré une néphroprotection bien menée, après évaluation de la balance bénéfice/risque.
Les formes, rares, avec insuffisance rénale rapidement progressive et prolifération extracapillaire > 50 % doivent être traitées comme les vascularites à ANCA : corticothérapie + bolus de cyclophosphamide. Les maladies néphrotiques pures, peu fréquentes, sans lésion histologique en dehors des dépôts d’IgA, requièrent une corticothérapie à 1 mg/kg/j à doses dégressives. Azathioprine, acide mycophénolique, rituximab, huiles de poisson, amygdalectomie systématique ne sont pas recommandés.
En cas de risque intermédiaire ou élevé (protéinurie > 0,5 g/j au diagnostic, plus ou moins associée à une HTA et une altération du DFGe), des mesures générales de néphroprotection sont recommandées. Une PA < 130/80 mmHg doit être obtenue avec des moyens non pharmacologiques (apports sodés limités à 6 g/j, activité physique, correction du surpoids, arrêt d’une intoxication éthylique chronique) et des thérapies médicamenteuses.
Les bloqueurs du système rénine-angiotensine sont indiqués – au premier rang desquels les IEC. Toutefois, un antagoniste des récepteurs de type 1 de l’angiotensine II (ARA II) peut être également prescrit (recommandation de niveau élevé lorsque la protéinurie initiale est > 1 g/24 h et suggéré si elle est située entre 0,5 et 1 g/24 h). L’association de ces 2 familles (double blocage) est possible, mais elle doit être initiée et surveillée par le néphrologue (risque accru d’instabilité de la fonction rénale).
Une restriction protidique modérée (0,6-0,8 g/kg/24 h) est préconisée, même si le niveau de preuve de cette recommandation est modeste.
Dans les formes les plus sévères, à risque évolutif élevé, des immunosuppresseurs sont envisageables. La corticothérapie a fait l’objet de plusieurs études contrôlées/randomisées et de méta-analyses, qui montrent un bénéfice en termes de réduction de la protéinurie et d’évolution de la fonction rénale ; cependant, dans ces essais les patients n’ont pas un contrôle tensionnel suffisant, et une faible fraction est traitée par IEC ou ARA II. Deux travaux récents ont remis en cause la place des corticoïdes au cours de la NIgA. Dans STOP-IGAN (étude randomisée contrôlée avec un schéma complexe vs placebo), une corticothérapie (si DFG > 60 mL/min/1,73 m2) ou le cyclophosphamide relayé par azathioprine (si DFG compris entre 30 et 60 mL/min/1,73 m2) était instauré lorsque la protéinurie restait > 0,75 g/j après 6 mois à 3 ans de néphroprotection maximale.8 Le critère de jugement principal a été atteint (rémission de la protéinurie, stabilité de la fonction rénale), mais aucun bénéfice n’a été retrouvé en termes d’évolution du DFG à 3 ans. Les auteurs concluent à l’absence d’effets significatifs au long cours sur la fonction rénale. Néanmoins, cette étude a de nombreux biais, notamment le fait que les patients pouvaient être inclus de nombreuses années après le diagnostic, sélectionnant des malades très stables. L’essai TESTING montre l’efficacité de la corticothérapie : régression de la protéinurie et gain de fonction rénale, versus placebo ;9 cependant, il a été interrompu précocement en raison d’un surcroît d’effets secondaires graves, essentiellement des infections, chez les patients traités.
Ainsi, les recommandations KDIGO ne proposent la cortico- thérapie (niveau de suggestion, 2C) qu’en cas de protéinurie persistante > 1 g/j malgré 3 à 6 mois de traitement antihypertenseur et antiprotéinurique (s’appuyant sur les IEC/ARA II si le DFG est > 50 mL/min/1,73 m2). Son utilisation est donc très débattue, mais elle n’est pas abandonnée. Elle est réservée à des patients jeunes, ayant une protéinurie > 1 g/j malgré une néphroprotection bien menée, après évaluation de la balance bénéfice/risque.
Les formes, rares, avec insuffisance rénale rapidement progressive et prolifération extracapillaire > 50 % doivent être traitées comme les vascularites à ANCA : corticothérapie + bolus de cyclophosphamide. Les maladies néphrotiques pures, peu fréquentes, sans lésion histologique en dehors des dépôts d’IgA, requièrent une corticothérapie à 1 mg/kg/j à doses dégressives. Azathioprine, acide mycophénolique, rituximab, huiles de poisson, amygdalectomie systématique ne sont pas recommandés.
Perspectives
Plusieurs traitements sont en développement. Une forme particulière de corticoïdes (budésonide), à libération digestive essentiellement iléale et à diffusion systémique limitée, pourrait cibler les anomalies inflammatoires muqueuses à l’origine de la synthèse d’IgA anormales. Dans une étude de phase II, ce médicament induit une réduction significative de la protéinurie et une moindre baisse du DFGe par rapport au placebo, avec un profil de tolérance excellent. Un essai de phase III est en cours.
L’impact du blocage d’un facteur de croissance des lymphocytes B, nommé BAFF, ou de la voie des lectines du complément sont également en cours d’investigation.
L’impact du blocage d’un facteur de croissance des lymphocytes B, nommé BAFF, ou de la voie des lectines du complément sont également en cours d’investigation.
Encadre
Que dire à vos patients
.Le suivi (analyse du sang et des urines), la prise régulière du traitement et le respect des consignes diététiques sont essentiels pour ralentir l’évolution.On ne peut pas en guérir complètement, mais on peut la contrôler.Elle a une composante génétique, mais le risque de la transmettre à son enfant est faible.Les associations de patients AIRG (https://www.airg-france.fr) et France Rein (https://www.francerein.org ; anciennement FNAIR) sont particulièrement actives, avec des réseaux régionaux.
références
1. Berthoux FC, Mohey H, Afiani A. Natural history of primary IgA nephropathy. Semin Nephrol 2008;28:4‑9.
2. Kiryluk K, Li Y, Scolari F, et al. Discovery of new risk loci for IgA nephro-pathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 2014;46:1187‑96.
3. Knoppova B, Reily C, Maillard N, et al. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front Immunol 2016; 7:117.
4. Launay P, Grossetête B, Arcos-Fajardo M, et al. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J Exp Med 2000;191:1999‑2009.
5. Trimarchi H, Barratt J, Cattran DC, et al. Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int 2017;91:1014‑21.
6. Berthoux F, Mohey H, Laurent B, Mariat C, Afiani A, Thibaudin L. Predicting the risk for dialysis or death in IgA nephropathy. J Am Soc Nephrol 2011;22:752‑61.
7. Radhakrishnan J, Cattran DC. The KDIGO practice guideline on glomerulonephritis : reading between the (guide)lines--application to the individual patient. Kidney Int 2012;82:840‑56.
8. Rauen T, Eitner F, Fitzner C, et al. Intensive Supportive Care plus Immuno-suppression in IgA Nephropathy. N Engl J Med 2015;373:2225‑36.
9. Lv J, Zhang H, Wong MG, et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Rando-mized Clinical Trial. JAMA 2017;318:432-42.
2. Kiryluk K, Li Y, Scolari F, et al. Discovery of new risk loci for IgA nephro-pathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 2014;46:1187‑96.
3. Knoppova B, Reily C, Maillard N, et al. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front Immunol 2016; 7:117.
4. Launay P, Grossetête B, Arcos-Fajardo M, et al. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J Exp Med 2000;191:1999‑2009.
5. Trimarchi H, Barratt J, Cattran DC, et al. Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int 2017;91:1014‑21.
6. Berthoux F, Mohey H, Laurent B, Mariat C, Afiani A, Thibaudin L. Predicting the risk for dialysis or death in IgA nephropathy. J Am Soc Nephrol 2011;22:752‑61.
7. Radhakrishnan J, Cattran DC. The KDIGO practice guideline on glomerulonephritis : reading between the (guide)lines--application to the individual patient. Kidney Int 2012;82:840‑56.
8. Rauen T, Eitner F, Fitzner C, et al. Intensive Supportive Care plus Immuno-suppression in IgA Nephropathy. N Engl J Med 2015;373:2225‑36.
9. Lv J, Zhang H, Wong MG, et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Rando-mized Clinical Trial. JAMA 2017;318:432-42.
 Encadrés
Encadrés