Cette maladie de présentation très hétérogène associe cytopénies, hépatosplénomégalie et atteinte osseuse, avec des formes très peu symptomatiques ou au contraire sévères. Le diagnostic est souvent fait avec retard alors qu’il faut débuter le traitement avant les complications potentiellement irréversibles.
La maladie de Gaucher est une maladie génétique rare, avec une prévalence estimée à 1/136 000 en France. Il s’agit néanmoins de la plus fréquente des maladies de surcharge lysosomale. C’est une maladie génétique à transmission autosomique récessive due à des mutations du gène GBA1, entraînant un déficit enzymatique en glucocérébrosidase.1 Son substrat, le glucosylcéramide, est issu de la dégradation des sphingolipides des membranes des globules rouges et globules blancs lorsqu’ils arrivent à leur fin de vie. Il s’accumule dans les lysosomes des macrophages, entraînant leur transformation en cellules de Gaucher. Ces cellules vont infiltrer le tissu splénique, hépatique, la moelle osseuse, et parfois les poumons ou le système nerveux central.
Des tableaux cliniques très divers
La présentation de la maladie de Gaucher est très hétérogène, allant de la forme asymptomatique jusqu’à la forme létale. Trois phénotypes et une forme fœtale sont classiquement distingués, cependant, la corrélation entre génotype et phénotype est parfois difficile à établir. Il semble plutôt exister un continuum entre ces différents phénotypes, fondé sur la présence et l’intensité des manifestations neurologiques.
Maladie de Gaucher de type 1
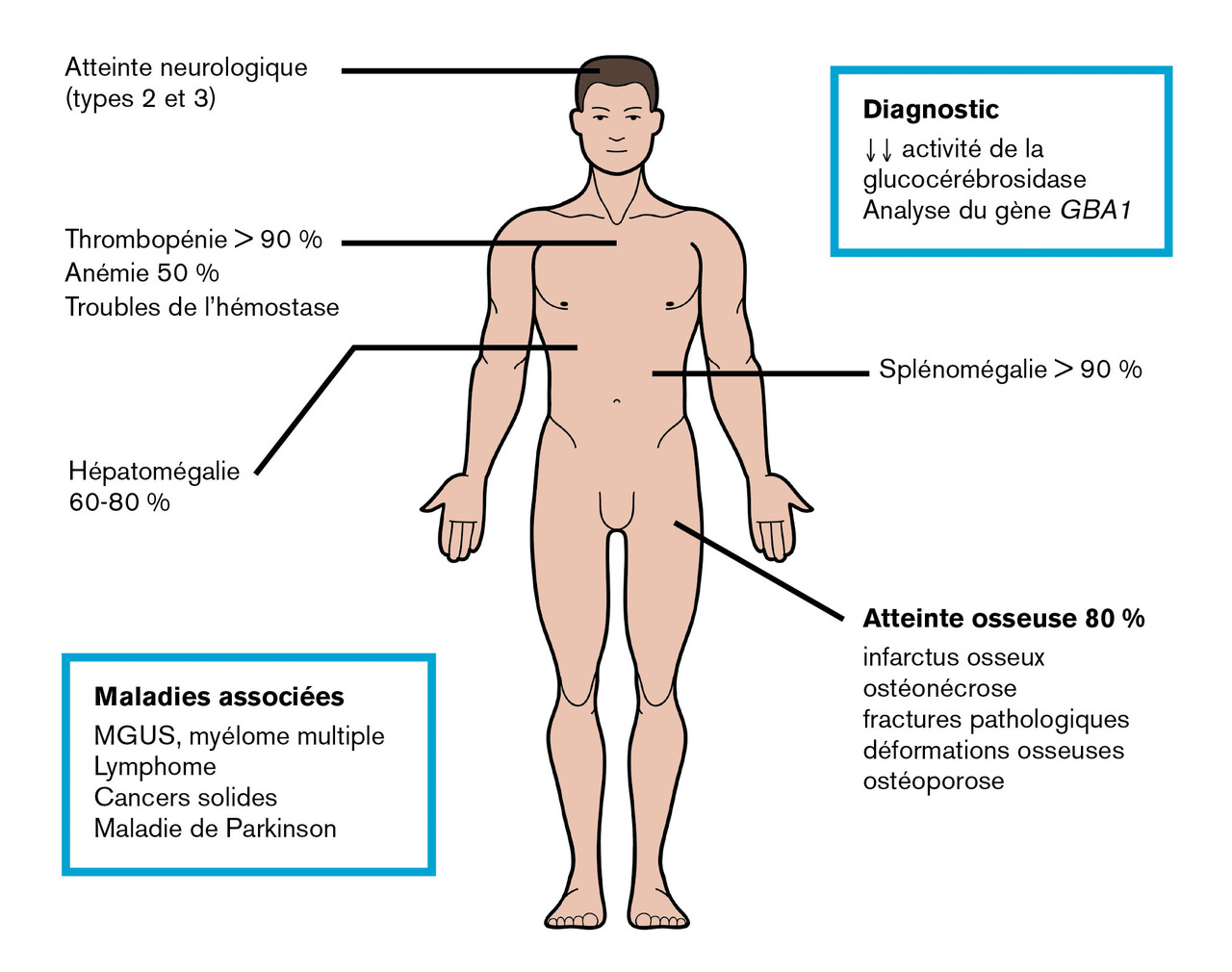
La maladie de Gaucher de type 1 (ORPHA77259) est la plus fréquente (95 % des cas). L’âge médian au diagnostic est de 22 ans (0-84 ans), mais il existe souvent un délai entre les premiers symptômes et le diagnostic. Sa présentation clinique est variable, allant de formes peu ou pas symptomatiques jusqu’aux formes très sévères (fig. 1 ).
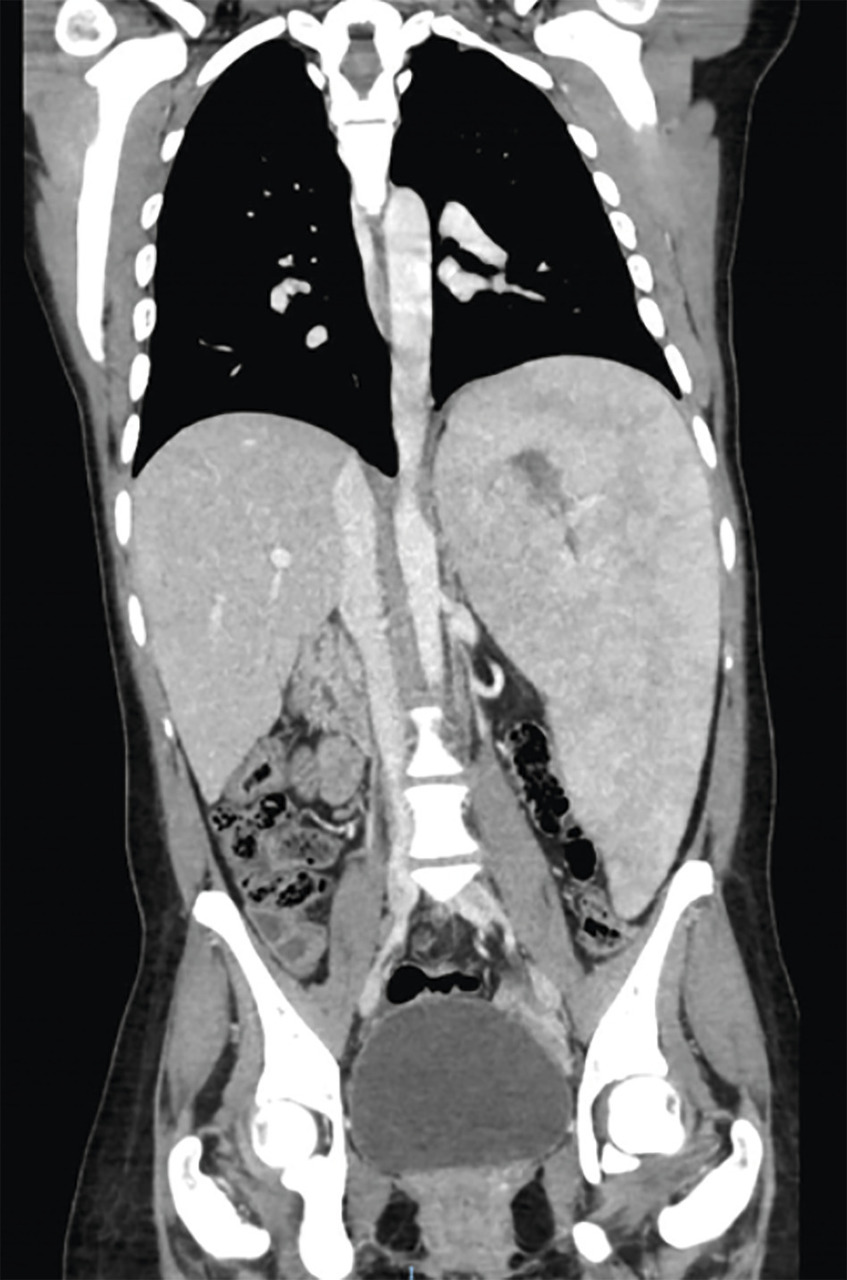
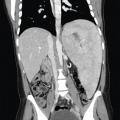
La splénomégalie est l’un des principaux signes cliniques, elle est présente chez plus de 90 % des patients, parfois massive et responsable d’une distension ou de douleurs abdominales. Il peut y avoir un hypersplénisme, participant à la thrombopénie. La splénomégalie peut se compliquer d’infarctus spléniques. Elle est souvent associée à une hépatomégalie, présente dans 60 à 80 % des cas (fig. 2 ). Des lésions focales de la rate ou du foie, appelées « gaucheromes », constituées d’amas de cellules de Gaucher peuvent être notées, pouvant poser le problème du diagnostic différentiel avec le carcinome hépatocellulaire, plus fréquent dans la maladie de Gaucher, ou avec une hémopathie.
L’atteinte osseuse (80 %) peut se manifester par des douleurs osseuses chroniques, ou par des événements ischémiques aigus : infarctus osseux et/ou ostéonécroses aseptiques. Ceux-ci sont dus à l’altération de la vascularisation osseuse liée à une infiltration par les cellules de Gaucher mais également à des mécanismes pro-inflammatoires et probablement vaso-occlusifs.
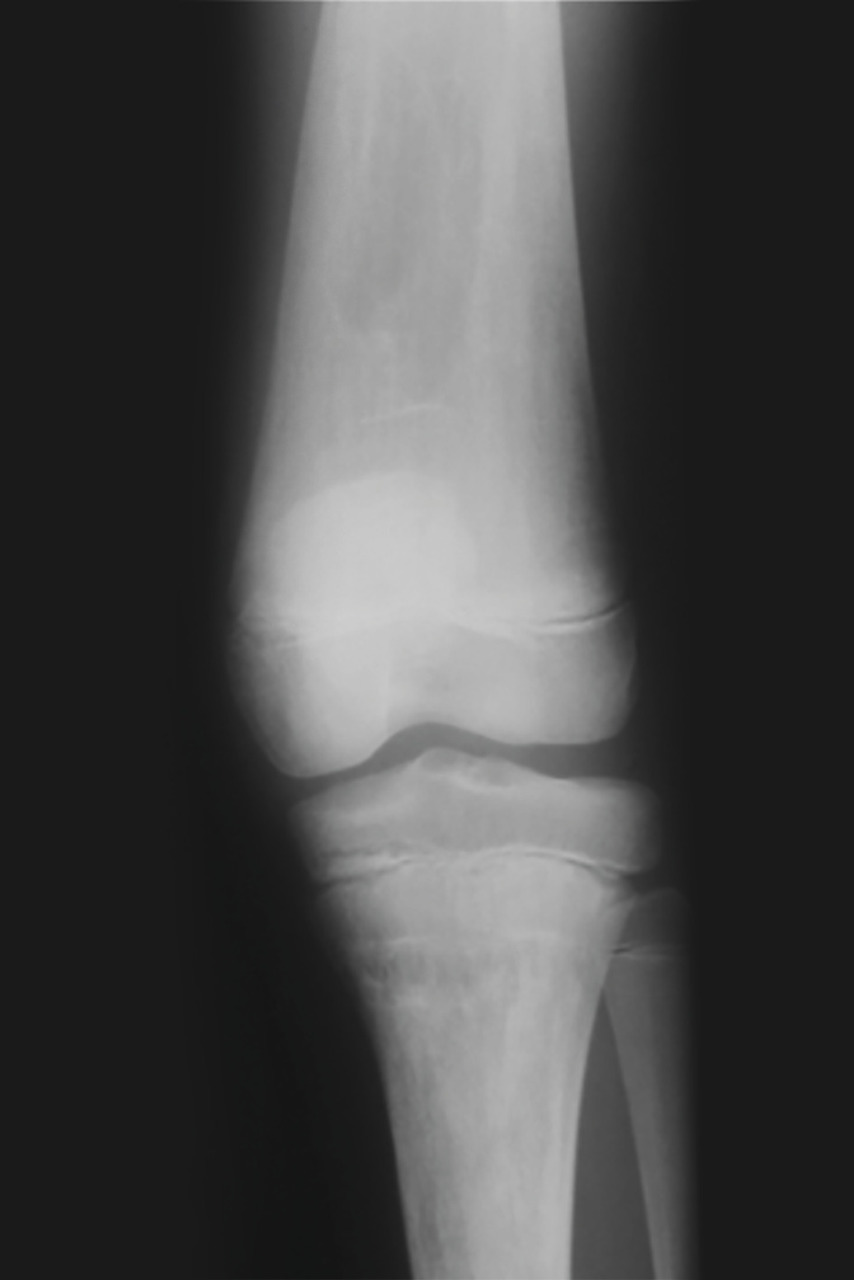
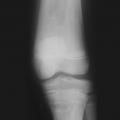
Les infarctus osseux peuvent se manifester par une fièvre modérée associée à un érythème et un œdème, localisés le plus souvent en regard des métaphyses et diaphyses des os longs. L’ostéonécrose aseptique, qui résulte de phénomènes identiques, est localisée aux épiphyses et atteint de manière préférentielle les têtes fémorales, les têtes humérales, et plus rarement les condyles fémoraux, les plateaux tibiaux, ou les corps vertébraux (vertebra plana). Les ostéonécroses épiphysaires peuvent conduire à une arthrose, justifiant à terme la pose d’une prothèse. La déformation des fémurs en flacon d’Erlenmeyer (fig. 3 ) correspond à un élargissement métaphysodiaphysaire des os longs, par défaut de remodelage osseux et de tubulation métaphysaire. Enfin, il existe une réduction de la densité minérale osseuse, avec un risque d’ostéoporose.
La splénomégalie est l’un des principaux signes cliniques, elle est présente chez plus de 90 % des patients, parfois massive et responsable d’une distension ou de douleurs abdominales. Il peut y avoir un hypersplénisme, participant à la thrombopénie. La splénomégalie peut se compliquer d’infarctus spléniques. Elle est souvent associée à une hépatomégalie, présente dans 60 à 80 % des cas (
L’atteinte osseuse (80 %) peut se manifester par des douleurs osseuses chroniques, ou par des événements ischémiques aigus : infarctus osseux et/ou ostéonécroses aseptiques. Ceux-ci sont dus à l’altération de la vascularisation osseuse liée à une infiltration par les cellules de Gaucher mais également à des mécanismes pro-inflammatoires et probablement vaso-occlusifs.
Les infarctus osseux peuvent se manifester par une fièvre modérée associée à un érythème et un œdème, localisés le plus souvent en regard des métaphyses et diaphyses des os longs. L’ostéonécrose aseptique, qui résulte de phénomènes identiques, est localisée aux épiphyses et atteint de manière préférentielle les têtes fémorales, les têtes humérales, et plus rarement les condyles fémoraux, les plateaux tibiaux, ou les corps vertébraux (vertebra plana). Les ostéonécroses épiphysaires peuvent conduire à une arthrose, justifiant à terme la pose d’une prothèse. La déformation des fémurs en flacon d’Erlenmeyer (
Maladie de Gaucher de type 2
La maladie de Gaucher de type 2 (ORPHA77260) est plus rare (< 1 %), et débute généralement chez le nourrisson de 3 à 6 mois avec une hépatosplénomégalie et une atteinte neurologique. L’association d’une raideur de nuque ou d’un opisthotonos, de signes bulbaires (notamment des troubles de la déglutition) et d’une paralysie oculomotrice horizontale est évocatrice. L’évolution est défavorable, avec un décès survenant avant la 3e année de vie, notamment du fait d’une atteinte pulmonaire infiltrative ou d’une apnée centrale.
Maladie de Gaucher de type 3
La maladie de Gaucher de type 3 (ORPHA77261) regroupe moins de 5 % des cas. Comme pour la maladie de Gaucher de type 1, son expression clinique est très hétérogène, avec un début en général avant l’âge de 2 ans. Certains cas plus lentement évolutifs sont diagnostiqués à l’âge adulte. En plus des atteintes du type 1 s’y ajoutent des signes neurologiques, allant d’une atteinte modérée avec une ophtalmoplégie horizontale supranucléaire (le patient n’arrivant pas à suivre un objet dans le plan horizontal, l’obligeant à tourner la tête) jusqu’aux formes plus invalidantes : syndrome cérébelleux et encéphalopathie progressive associant épilepsie myoclonique, myoclonies non épileptiques, spasticité, troubles du comportement, voire démence. Les signes neurologiques peuvent survenir plusieurs années avant les autres manifestations cliniques. Une infiltration interstitielle pulmonaire, une cyphose souvent invalidante et de volumineuses adénopathies généralement abdominales sont parfois retrouvées. Des calcifications valvulaires aortiques et coronariennes, une opacité de la cornée, et une hydrocéphalie sont caractéristiques du type 3C ayant le génotype p.Asp448his à l’état homozygote.
Forme fœtale
La forme fœtale (anasarque fœtoplacentaire, ichtyose, arthrogrypose, etc.) de la maladie de Gaucher est exceptionnelle, responsable d’un décès in utero ou juste après la naissance.2
Investigations paracliniques
La thrombopénie est fréquente (90 % des cas) et de degré variable (< 60 G/L dans 26 % des cas). Une anémie est présente dans 56 % des cas, souvent modérée, avec un taux d’hémoglobine rarement en dessous de 9 g/dL. Des leucopénies peuvent être observées mais sont rares. Le mécanisme des cytopénies est attribué à l’hypersplénisme et à l’infiltration médullaire, mais un impact direct du déficit enzymatique sur les cellules souches hématopoïétiques a également été décrit.
La protéine C-réactive peut être élevée mais uniquement en cas d’infarctus osseux ou lors des complications infectieuses. Une hyperferritinémie est fréquemment retrouvée (> 85 %), alors que le fer sérique et le coefficient de saturation de la transferrine restent normaux.
Une hypergammaglobulinémie polyclonale est retrouvée dans 25 à 91 % des cas en fonction des séries. Par ailleurs, il y a parfois un ou plusieurs pics monoclonaux, dont la prévalence est également augmentée au cours de la maladie de Gaucher (entre 1 et 35 % selon les séries).3 Il s’agit le plus souvent de gammapathies monoclonales de signification indéterminée. Certains auteurs suggèrent que certains lysolipides (notamment la glucosylsphingosine) pourraient être l’antigène reconnu par ce composant monoclonal.
En ce qui concerne les imageries, l’échographie abdominale reste largement utilisée en première intention, car facilement disponible. Néanmoins, l’imagerie par résonance magnétique (IRM) abdominale est l’examen de choix pour évaluer les dimensions de la rate et du foie, ainsi que leur morphologie, et pour évaluer les nodules en rapport avec des « gaucheromes ». L’IRM est également l’examen de choix pour évaluer l’atteinte osseuse, pour quantifier l’infiltration osseuse par les cellules de Gaucher, l’extension des lésions, leur caractère aigu ou ancien, et pour le suivi des patients. Les radiographies osseuses sont utiles pour toute localisation symptomatique et le suivi des ostéonécroses. Les lésions rencontrées sont multiples : déformation fémorale en flacon d’Erlenmeyer (fig. 3 ), lésions lytiques, amincissement de la corticale, lésions d’ostéo-condensation, fractures. Enfin, l’ostéodensitométrie permet de détecter une ostéopénie ou une ostéoporose, fréquentes dans la maladie de Gaucher.
La protéine C-réactive peut être élevée mais uniquement en cas d’infarctus osseux ou lors des complications infectieuses. Une hyperferritinémie est fréquemment retrouvée (> 85 %), alors que le fer sérique et le coefficient de saturation de la transferrine restent normaux.
Une hypergammaglobulinémie polyclonale est retrouvée dans 25 à 91 % des cas en fonction des séries. Par ailleurs, il y a parfois un ou plusieurs pics monoclonaux, dont la prévalence est également augmentée au cours de la maladie de Gaucher (entre 1 et 35 % selon les séries).3 Il s’agit le plus souvent de gammapathies monoclonales de signification indéterminée. Certains auteurs suggèrent que certains lysolipides (notamment la glucosylsphingosine) pourraient être l’antigène reconnu par ce composant monoclonal.
En ce qui concerne les imageries, l’échographie abdominale reste largement utilisée en première intention, car facilement disponible. Néanmoins, l’imagerie par résonance magnétique (IRM) abdominale est l’examen de choix pour évaluer les dimensions de la rate et du foie, ainsi que leur morphologie, et pour évaluer les nodules en rapport avec des « gaucheromes ». L’IRM est également l’examen de choix pour évaluer l’atteinte osseuse, pour quantifier l’infiltration osseuse par les cellules de Gaucher, l’extension des lésions, leur caractère aigu ou ancien, et pour le suivi des patients. Les radiographies osseuses sont utiles pour toute localisation symptomatique et le suivi des ostéonécroses. Les lésions rencontrées sont multiples : déformation fémorale en flacon d’Erlenmeyer (
Confirmation diagnostique
Le diagnostic de maladie de Gaucher doit être confirmé en mesurant l’activité de la glucocérébrosidase dans les leucocytes, les cellules mononucléées ou les fibroblastes. La mise en évidence d’un déficit enzymatique (10-15 % de l’activité normale) est l’examen de référence.
L’analyse du gène de la glucocérébrosidase (GBA1) situé sur le chromosome 1 doit ensuite être réalisée afin d’identifier les variants pathogènes des deux allèles. Actuellement, plus de 300 mutations ont été décrites. Certaines sont très fréquentes, comme la mutation p.Asn409Ser, et p.Leu483Pro. Le génotype peut, dans certains cas, apporter des informations sur le pronostic ainsi que sur le phénotype.
L’analyse du gène de la glucocérébrosidase (GBA1) situé sur le chromosome 1 doit ensuite être réalisée afin d’identifier les variants pathogènes des deux allèles. Actuellement, plus de 300 mutations ont été décrites. Certaines sont très fréquentes, comme la mutation p.Asn409Ser, et p.Leu483Pro. Le génotype peut, dans certains cas, apporter des informations sur le pronostic ainsi que sur le phénotype.
Maladies associées
Chez les patients atteints de maladie de Gaucher de type 1, le risque de maladie de Parkinson est augmenté de 6 à 17 fois comparativement à la population générale, avec des formes en général plus précoces.4 Parallèlement, il existe une forte association entre les mutations du gène GBA1, hétérozygotes ou homozygotes, et la maladie de Parkinson, surtout avec les mutations associées à un phénotype neurologique de maladie de Gaucher, notamment p.Leu483Pro. L’efficacité du traitement spécifique de la maladie de Gaucher sur l’incidence de la maladie de Parkinson n’est néanmoins pas connue.
Par ailleurs, le risque de certains cancers solides comme le carcinome hépatocellulaire, le mélanome, et le cancer du pancréas, semble augmenté chez les patients atteints de maladie de Gaucher.
Enfin, l’incidence du myélome multiple est augmentée, avec un risque relatif estimé entre 1,3 à 51 comparativement à la population générale.
Par ailleurs, le risque de certains cancers solides comme le carcinome hépatocellulaire, le mélanome, et le cancer du pancréas, semble augmenté chez les patients atteints de maladie de Gaucher.
Enfin, l’incidence du myélome multiple est augmentée, avec un risque relatif estimé entre 1,3 à 51 comparativement à la population générale.
Quand évoquer la maladie de Gaucher ?
Malgré la disponibilité de tests diagnostiques sensibles et peu invasifs, l’absence de spécificité des symptômes et la rareté de la maladie ont pour conséquence des retards diagnostiques (49 ± 124 mois en moyenne) pouvant entraîner des complications irréversibles (ostéonécroses aseptiques, fractures pathologiques, hépatopathies ou hémorragies sévères), pourtant évitables avec les traitements spécifiques. Un groupe international d’experts a publié une liste de critères devant faire évoquer le diagnostic (v. tableau ) : splénomégalie, thrombopénie (en particulier pendant la grossesse), manifestations osseuses, hyperferritinémie, hépatomégalie, gammapathie monoclonale ou polyclonale, antécédents familiaux de maladie de Gaucher et origine juive ashkénaze.5 Le clinicien doit surtout évoquer la maladie de Gaucher en cas d’association de deux ou de plusieurs de ces symptômes, en l’absence de diagnostic différentiel. Une association avec une maladie de Parkinson, surtout précoce, peut également être évocatrices.
Comment la prendre en charge ?
En France, le Centre de référence des maladies lysosomales et le Comité d’évaluation du traitement de la maladie de Gaucher ont rédigé un protocole national de diagnostic et de soins, édité par la Haute Autorité de santé, permettant d’harmoniser les pratiques en termes de diagnostic, de suivi et de traitement.6
Le traitement spécifique doit être discuté, de préférence au cours d’une réunion de concertation pluridisciplinaire, en particulier au cours des formes asymptomatiques ou peu symptomatiques pour lesquelles leur indication n’est pas systématique. Il existe deux types de traitements spécifiques : l’enzymothérapie substitutive et les réducteurs de substrat.
L’enzymothérapie substitutive (imiglucérase, vélaglucérase) compense la diminution de l’activité de la glucocérébrosidase et s’administre par voie intraveineuse tous les 14 jours, parfois à domicile. Son efficacité a été démontrée sur l’amélioration des cytopénies, des organomégalies, des douleurs osseuses, de la densité osseuse mais également sur la qualité de vie. Ces traitements sont en général bien tolérés. Actuellement, il n’y a pas d’indication à l’enzymothérapie substitutive dans la maladie de Gaucher de type 2, en l’absence d’efficacité prouvée sur l’atteinte neurologique.
Les réducteurs de substrat, comprenant l’éliglustat et le miglustat, ont pour avantage une administration orale. L’éliglustat est un analogue de la céramide inhibant la glucosylcéramide synthase, de façon plus spécifique et plus puissante que le miglustat, qui n’est pratiquement plus utilisé en raison de sa mauvaise tolérance digestive et de sa moindre efficacité. L’éliglustat a été évalué uniquement dans la maladie de Gaucher de type 1, et a montré son efficacité sur les cytopénies, les organomégalies ainsi que sur la densité osseuse. Néanmoins se pose le problème d’interactions médicamenteuses, car l’éliglustat est métabolisé par le cytochrome P450, ce qui nécessite de déterminer le statut métaboliseur des patients par un génotypage du CYP2D6.
Aujourd’hui, la splénectomie n’a plus d’indication à l’heure des traitements spécifiques, car elle augmente le risque infectieux ainsi que les risques de complications osseuses, de fibrose hépatique et d’hypertension artérielle pulmonaire.
Les patients doivent être suivis régulièrement sur les plans clinique, biologique et radiologique. La prise en charge est organisée en France grâce aux centres de référence et de compétence des maladies lysosomales appartenant à la filière de santé maladies rares G2M : maladies héréditaires du métabolisme G2M (www.filiere-g2m.fr). Les patients doivent bénéficier d’une éducation thérapeutique sur leur maladie et être informés de l’existence d’associations de patients (association Vaincre les maladies lysosomales [VML], www.vml-asso.org), ce qui contribue à une meilleure prise en charge globale de cette maladie rare.
Le traitement spécifique doit être discuté, de préférence au cours d’une réunion de concertation pluridisciplinaire, en particulier au cours des formes asymptomatiques ou peu symptomatiques pour lesquelles leur indication n’est pas systématique. Il existe deux types de traitements spécifiques : l’enzymothérapie substitutive et les réducteurs de substrat.
L’enzymothérapie substitutive (imiglucérase, vélaglucérase) compense la diminution de l’activité de la glucocérébrosidase et s’administre par voie intraveineuse tous les 14 jours, parfois à domicile. Son efficacité a été démontrée sur l’amélioration des cytopénies, des organomégalies, des douleurs osseuses, de la densité osseuse mais également sur la qualité de vie. Ces traitements sont en général bien tolérés. Actuellement, il n’y a pas d’indication à l’enzymothérapie substitutive dans la maladie de Gaucher de type 2, en l’absence d’efficacité prouvée sur l’atteinte neurologique.
Les réducteurs de substrat, comprenant l’éliglustat et le miglustat, ont pour avantage une administration orale. L’éliglustat est un analogue de la céramide inhibant la glucosylcéramide synthase, de façon plus spécifique et plus puissante que le miglustat, qui n’est pratiquement plus utilisé en raison de sa mauvaise tolérance digestive et de sa moindre efficacité. L’éliglustat a été évalué uniquement dans la maladie de Gaucher de type 1, et a montré son efficacité sur les cytopénies, les organomégalies ainsi que sur la densité osseuse. Néanmoins se pose le problème d’interactions médicamenteuses, car l’éliglustat est métabolisé par le cytochrome P450, ce qui nécessite de déterminer le statut métaboliseur des patients par un génotypage du CYP2D6.
Aujourd’hui, la splénectomie n’a plus d’indication à l’heure des traitements spécifiques, car elle augmente le risque infectieux ainsi que les risques de complications osseuses, de fibrose hépatique et d’hypertension artérielle pulmonaire.
Les patients doivent être suivis régulièrement sur les plans clinique, biologique et radiologique. La prise en charge est organisée en France grâce aux centres de référence et de compétence des maladies lysosomales appartenant à la filière de santé maladies rares G2M : maladies héréditaires du métabolisme G2M (www.filiere-g2m.fr). Les patients doivent bénéficier d’une éducation thérapeutique sur leur maladie et être informés de l’existence d’associations de patients (association Vaincre les maladies lysosomales [VML], www.vml-asso.org), ce qui contribue à une meilleure prise en charge globale de cette maladie rare.
Références
1. Brady RO, Kanfer JN, Shapiro D. Metabolism of Glucocerebrosides. II. Evidence of an Enzymatic Deficiency in Gaucher’s Disease. Biochem. Biophys Res Commun 1965;18:221-5.
2. Mignot C, Gelot A, Bessières B, et al. Perinatal-lethal Gaucher disease. Am J Med Genet A 2003; 120A:338-44.
3. Arends M, van Dussen L, Biegstraaten M, Hollak CEM. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol 2013;161:832-42.
4. Rosenbloom B, Balwani M, Bronstein JM, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher registry. Blood Cells Mol Dis 2011;46:95-102.
5. Mehta A, Kuter DJ, Salek SS, et al. Presenting signs and patient co-variables in Gaucher disease: outcome of the Gaucher Early Diagnosis Consensus (GED-C) Delphi initiative. Intern Med J 2019;49:578-91.
6. Haute Autorité de santé. Maladie de Gaucher. Guide Maladie chronique, HAS 2015. www.has-sante.fr ou https://bit.ly/2UmTuqG
7. Sidransky, E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 2004;83:6-15.
8. Mehta A, Belmatoug N, Bembi B, et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol Genet Metab 2017;122:122-9.
2. Mignot C, Gelot A, Bessières B, et al. Perinatal-lethal Gaucher disease. Am J Med Genet A 2003; 120A:338-44.
3. Arends M, van Dussen L, Biegstraaten M, Hollak CEM. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol 2013;161:832-42.
4. Rosenbloom B, Balwani M, Bronstein JM, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher registry. Blood Cells Mol Dis 2011;46:95-102.
5. Mehta A, Kuter DJ, Salek SS, et al. Presenting signs and patient co-variables in Gaucher disease: outcome of the Gaucher Early Diagnosis Consensus (GED-C) Delphi initiative. Intern Med J 2019;49:578-91.
6. Haute Autorité de santé. Maladie de Gaucher. Guide Maladie chronique, HAS 2015. www.has-sante.fr ou https://bit.ly/2UmTuqG
7. Sidransky, E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 2004;83:6-15.
8. Mehta A, Belmatoug N, Bembi B, et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol Genet Metab 2017;122:122-9.
Dans cet article

Résumé
La maladie de Gaucher est une maladie lysosomale génétique rare à transmission autosomique récessive, due à un déficit enzymatique en glucocérébrosidase. La présentation clinique est très hétérogène, allant de formes asymptomatiques ou très peu symptomatiques à des formes sévères. Elle associe des cytopénies, une hépatosplénomégalie et une atteinte osseuse (ostéonécroses aseptiques, infarctus osseux, fractures pathologiques et ostéoporose). Des atteintes neurologiques potentiellement sévères peuvent se voir dans les maladies de Gaucher de type 2 et 3. Le type 1 est associé à un risque accru de maladie de Parkinson, de certains cancers solides, et d’hémopathies dont le myélome multiple. Le diagnostic est souvent fait avec retard, l’enjeu est donc celui d’une meilleure connaissance des symptômes devant faire évoquer la maladie, afin d’initier un traitement avant les complications potentiellement irréversibles.