Hématologie. La population des patients atteints de cette maladie vieillit, et de nombreuses comorbidités viennent compliquer leur prise en charge. Le risque hémorragique doit être pris en compte de manière spécifique lors de décisions thérapeutiques.
Mise au point
La maladie de Willebrand est une maladie hémorragique héréditaire qui résulte d’une anomalie du facteur Willebrand, considérée comme la plus fréquente ; ses formes symptomatiques ne touchent cependant qu’environ 1 individu sur 10 000, il s’agit donc d’une maladie rare.
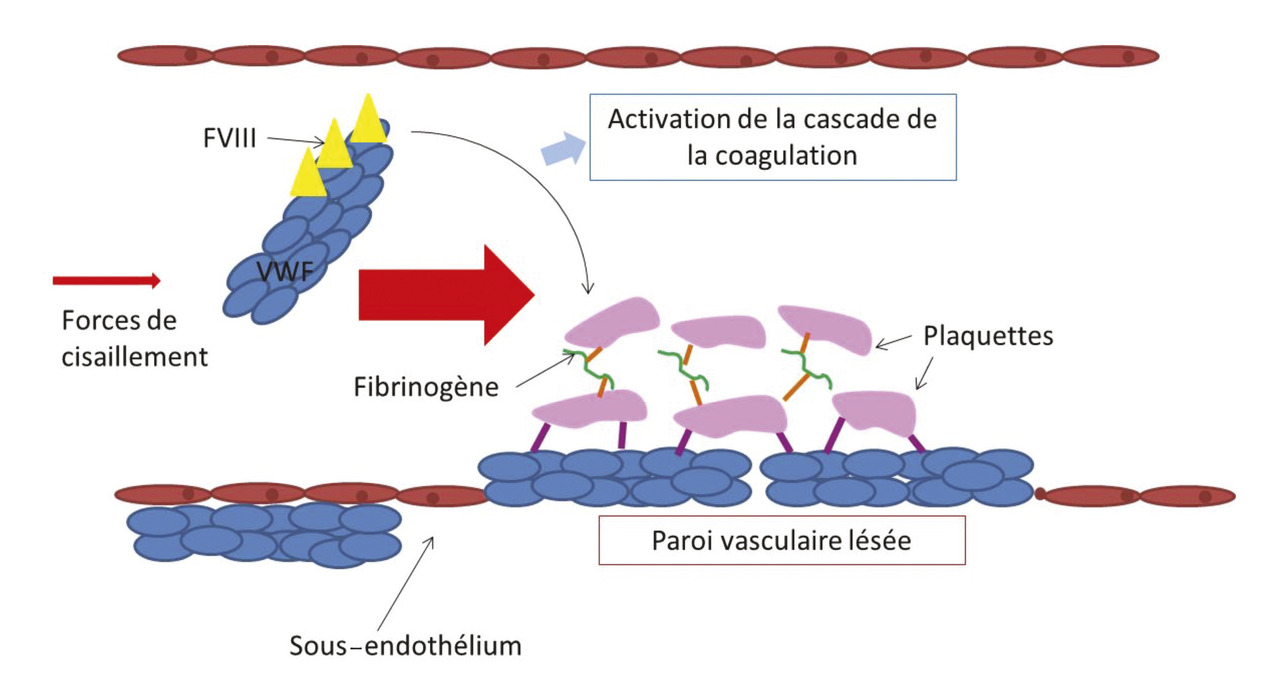
Le facteur Willebrand (von Willebrand factor [VWF]) est une glycoprotéine synthétisée dans les cellules endothéliales et les mégacaryocytes. Sa structure multimérique lui confère deux grandes fonctions : il permet l’adhésion des plaquettes à la paroi vasculaire lésée et facilite l’agrégation des plaquettes entre elles ; il se lie au facteur VIII de la coagulation pour en assurer le transport et sa protection contre une protéolyse plasmatique, le facteur VIII libre ayant une demi-vie courte de 2 heures ; quand il est lié au facteur Willebrand, cette demi-vie dépasse 12 à 15 heures. Le facteur Willebrand est sensible aux forces de cisaillement qui se majorent lors d’une brèche vasculaire entraînant au niveau de la paroi lésée à la fois l’activation de l’hémostase primaire et l’accélération de la cascade de la coagulation par la libération du facteur VIII( v. figure) .
La maladie de Willebrand, dont la définition a été précisée en 2006, est due à un défaut génétique du gène VWF (ou tout autre gène) aboutissant soit à un déficit quantitatif, soit à un déficit qualitatif du facteur Willebrand.1 Cette anomalie génétique est transmise le plus souvent de façon autosomique dominante. Ainsi, on distingue trois types de maladie de Willebrand : le type 1 correspond à un déficit quantitatif partiel en facteur Willebrand, le type 3 (forme récessive) à un déficit quasi-total en facteur Willebrand, et le type 2 à un déficit qualitatif touchant soit l’interaction du facteur Willebrand avec les plaquettes (et/ou le sous-endothélium), soit l’interaction avec le facteur VIII( v. tableau) .
Ces différents types de la maladie sont à l’origine de tableaux cliniques très hétérogènes et impliquent une prise en charge thérapeutique variable et spécifique. Grâce aux différents progrès de la médecine et de la prise en charge, l’espérance de vie des patients atteints de maladie de Willebrand tend à s’approcher de celle de la population générale. Ainsi cette population de patients vieillit et peut avoir un certain nombre de comorbidités ou de situations dont la fréquence augmente avec l’âge.
L’objectif de cet article est de faire une synthèse des propositions thérapeutiques actuelles pour prendre en charge les comorbidités les plus fréquentes chez les sujets âgés atteints de maladie de Willebrand, dont le risque hémorragique doit être pris en compte de manière spécifique. Il ne s’agit donc ni de recommandations ni de consensus d’experts.
Sont exclus de ce propos les patients ayant un syndrome de Willebrand acquis dont la physiopathologie est différente, conséquence d’un processus sous-jacent.
La maladie de Willebrand est une maladie hémorragique héréditaire qui résulte d’une anomalie du facteur Willebrand, considérée comme la plus fréquente ; ses formes symptomatiques ne touchent cependant qu’environ 1 individu sur 10 000, il s’agit donc d’une maladie rare.
Le facteur Willebrand (von Willebrand factor [VWF]) est une glycoprotéine synthétisée dans les cellules endothéliales et les mégacaryocytes. Sa structure multimérique lui confère deux grandes fonctions : il permet l’adhésion des plaquettes à la paroi vasculaire lésée et facilite l’agrégation des plaquettes entre elles ; il se lie au facteur VIII de la coagulation pour en assurer le transport et sa protection contre une protéolyse plasmatique, le facteur VIII libre ayant une demi-vie courte de 2 heures ; quand il est lié au facteur Willebrand, cette demi-vie dépasse 12 à 15 heures. Le facteur Willebrand est sensible aux forces de cisaillement qui se majorent lors d’une brèche vasculaire entraînant au niveau de la paroi lésée à la fois l’activation de l’hémostase primaire et l’accélération de la cascade de la coagulation par la libération du facteur VIII
La maladie de Willebrand, dont la définition a été précisée en 2006, est due à un défaut génétique du gène VWF (ou tout autre gène) aboutissant soit à un déficit quantitatif, soit à un déficit qualitatif du facteur Willebrand.1 Cette anomalie génétique est transmise le plus souvent de façon autosomique dominante. Ainsi, on distingue trois types de maladie de Willebrand : le type 1 correspond à un déficit quantitatif partiel en facteur Willebrand, le type 3 (forme récessive) à un déficit quasi-total en facteur Willebrand, et le type 2 à un déficit qualitatif touchant soit l’interaction du facteur Willebrand avec les plaquettes (et/ou le sous-endothélium), soit l’interaction avec le facteur VIII
Ces différents types de la maladie sont à l’origine de tableaux cliniques très hétérogènes et impliquent une prise en charge thérapeutique variable et spécifique. Grâce aux différents progrès de la médecine et de la prise en charge, l’espérance de vie des patients atteints de maladie de Willebrand tend à s’approcher de celle de la population générale. Ainsi cette population de patients vieillit et peut avoir un certain nombre de comorbidités ou de situations dont la fréquence augmente avec l’âge.
L’objectif de cet article est de faire une synthèse des propositions thérapeutiques actuelles pour prendre en charge les comorbidités les plus fréquentes chez les sujets âgés atteints de maladie de Willebrand, dont le risque hémorragique doit être pris en compte de manière spécifique. Il ne s’agit donc ni de recommandations ni de consensus d’experts.
Sont exclus de ce propos les patients ayant un syndrome de Willebrand acquis dont la physiopathologie est différente, conséquence d’un processus sous-jacent.
Hémorragie
Physiologiquement, les taux de facteur Willebrand augmentent avec l’âge.2 Chez les sujets atteints de la maladie de Willebrand de type 1 modérée, on assiste ainsi à une augmentation progressive des taux de facteur Willebrand avec l’âge, aussi bien ceux de l’antigène (VWF:Ag) que ceux de l’activité (le plus souvent activité cofacteur de la ristocétine, VWF:RCo), permettant d’expliquer que la fréquence de la symptomatologie hémorragique de type cutanéo-muqueux diminue. Ce n’est pas le cas pour les sujets atteints du type 3 qui ont un déficit complet en facteur Willebrand. De même, chez les patients atteints d’une maladie de Willebrand de type 2, le déficit qualitatif persiste même si le taux de VWF:Ag circulant est augmenté : la dissociation entre l’antigène et l’activité persiste, et témoigne du déficit fonctionnel en facteur Willebrand. Le taux de facteur VIII suit l’ascension de celui du VWF:Ag dans la mesure où il circule lié au facteur Willebrand.
Il est néanmoins intéressant de noter qu’au cours du vieillissement la localisation de la symptomatologie hémorragique change : les hémorragies gastro-intestinales et les hématomes spontanés sont plus fréquents chez les sujets âgés comparés aux sujets jeunes.2, 3
Le traitement de la maladie de Willebrand, dans ses grandes lignes, comprend deux types de traitement spécifique qui peuvent s’accompagner de traitements adjuvants :
– le premier consiste à utiliser la desmopressine en s’assurant au préalable de la réponse à ce médicament par une épreuve thérapeutique, cela en l’absence de contre-indication. Celles-ci sont : l’âge supérieur à 70 ans (et inférieur à 2 ans), l’hypertension artérielle, le type 2B de la maladie de Willebrand. La population gériatrique ne peut ainsi pas ou plus bénéficier de ce traitement ;4
– le second repose sur une substitution par des concentrés de facteur Willebrand, associés dans certains cas à des concentrés de facteur VIII, tous deux médicaments dérivés du sang. Ceux-ci ne sont distribués que par une pharmacie hospitalière selon un protocole établi par un médecin senior en fonction de la symptomatologie du patient, du contexte médical ou chirurgical et du type de maladie de Willebrand. Les posologies sont calculées en fonction du poids du patient, 1 UI/kg augmentant d’environ 2 % le taux de ces facteurs.
Dans tous les cas, un suivi biologique rapproché doit être effectué, comprenant une mesure de l’activité du facteur Willebrand, le plus souvent activité cofacteur de la ristocétine (VWF:RCo), du VWF:Ag et du facteur VIII, pour s’assurer de l’efficacité et ajuster les posologies en cas de substitution.
Enfin, les traitements adjuvants chez le patient âgé associent des antifibrinolytiques (acide tranexamique) et les compresses hémostatiques.
Il est néanmoins intéressant de noter qu’au cours du vieillissement la localisation de la symptomatologie hémorragique change : les hémorragies gastro-intestinales et les hématomes spontanés sont plus fréquents chez les sujets âgés comparés aux sujets jeunes.2, 3
Le traitement de la maladie de Willebrand, dans ses grandes lignes, comprend deux types de traitement spécifique qui peuvent s’accompagner de traitements adjuvants :
– le premier consiste à utiliser la desmopressine en s’assurant au préalable de la réponse à ce médicament par une épreuve thérapeutique, cela en l’absence de contre-indication. Celles-ci sont : l’âge supérieur à 70 ans (et inférieur à 2 ans), l’hypertension artérielle, le type 2B de la maladie de Willebrand. La population gériatrique ne peut ainsi pas ou plus bénéficier de ce traitement ;4
– le second repose sur une substitution par des concentrés de facteur Willebrand, associés dans certains cas à des concentrés de facteur VIII, tous deux médicaments dérivés du sang. Ceux-ci ne sont distribués que par une pharmacie hospitalière selon un protocole établi par un médecin senior en fonction de la symptomatologie du patient, du contexte médical ou chirurgical et du type de maladie de Willebrand. Les posologies sont calculées en fonction du poids du patient, 1 UI/kg augmentant d’environ 2 % le taux de ces facteurs.
Dans tous les cas, un suivi biologique rapproché doit être effectué, comprenant une mesure de l’activité du facteur Willebrand, le plus souvent activité cofacteur de la ristocétine (VWF:RCo), du VWF:Ag et du facteur VIII, pour s’assurer de l’efficacité et ajuster les posologies en cas de substitution.
Enfin, les traitements adjuvants chez le patient âgé associent des antifibrinolytiques (acide tranexamique) et les compresses hémostatiques.
Pathologies cardiovasculaires
Hypertension artérielle
L’hypertension artérielle est connue pour être un facteur de risque important des maladies cardiovasculaires. Une étude a permis de suivre pendant 2 ans 258 patients ayant une hémophilie ou une maladie de Willebrand et une hypertension artérielle.5 La moyenne d’âge était de 60 ans (pas de détails concernant les patients > 75 ans), âges extrêmes : 24-92 ans). La plupart étaient atteints d’une maladie de Willebrand de type 1 (n = 190 ; 73,4 %). La majorité de ces patients avaient un traitement antihypertenseur, soit 84,5 %. À noter que 22,1 % des patients (hémophilie A et maladie de Willebrand confondues) avaient une coronaropathie.
Néanmoins, plusieurs études ont montré que les patients atteints de maladies hémorragiques ont un risque significativement inférieur de développer une artériopathie comparativement à une population contrôle à condition de ne pas avoir de facteurs de risque surajoutés tels qu’une hypercholestérolémie, une obésité, ou un tabagisme.6, 7
Cependant, les indications thérapeutiques des sociétés savantes de cardiologie en cas d’hypertension artérielle doivent s’appliquer aussi aux patients atteints d’une maladie de Willebrand.
Néanmoins, plusieurs études ont montré que les patients atteints de maladies hémorragiques ont un risque significativement inférieur de développer une artériopathie comparativement à une population contrôle à condition de ne pas avoir de facteurs de risque surajoutés tels qu’une hypercholestérolémie, une obésité, ou un tabagisme.6, 7
Cependant, les indications thérapeutiques des sociétés savantes de cardiologie en cas d’hypertension artérielle doivent s’appliquer aussi aux patients atteints d’une maladie de Willebrand.
Coronaropathies, artériopathies
Les recommandations internationales pour la prise en charge des maladies coronaires aiguës sont largement diffusées pour la population générale par les sociétés savantes de cardiologie.8-10 Plusieurs études ont été menées auprès de la population générale âgée ayant une coronaropathie. L’un des risques majeurs pour ce type de patients de plus de 75 ans est l’augmentation du risque hémorragique par le traitement antiagrégant.11, 12 Il est apparu pour ces patients âgés, atteints d’une pathologie coronarienne, la nécessité d’opter pour une stratégie thérapeutique limitant ce risque hémorragique.
En cas d’angioplastie, les stents actifs (drug eluting stent [DES]) récents avec des polymères bioabsorbables permettent de diminuer la durée de la double antiagrégation plaquettaire à 1 mois comme cela est préconisé en cas de pose d’un stent nu (bare metal stent [BMS])12. Il a été démontré que les stents actifs présentent moins de risque de thrombose que les stents nus.13, 14 Ainsi, il paraîtrait intéressant d’associer les stents actifs à polymères résorbables et une double antiagrégation plaquettaire pour les patients âgés ayant une maladie de Willebrand et, dans une plus large mesure, tout patient ayant une maladie hémorragique. Néanmoins, à notre connaissance, aucune publication n’est parue à ce jour.
À l’heure actuelle, selon les données publiées, il faut pour les patients ayant une maladie de Willebrand : 4, 15-17
– évaluer précisément le phénotype hémorragique du patient par un interrogatoire exhaustif des antécédents hémorragiques ; 4, 15-17
– lors d’une coronarographie et/ou d’une angioplastie, privilégier la voie d’abord radiale car elle permet une compression du point de ponction plus facile qu’une voie d’abord fémorale et limite le risque hémorragique ; 4, 15-17
– en cas de procédures invasives, effectuer le geste sous couvert d’un traitement substitutif de concentrés de facteur Willebrand associé ou non au facteur VIII, 30 à 60 minutes avant et 24 à 48 heures après. La cible thérapeutique de la substitution est située au pic entre 80 et 100 % de VWF:RCo et du facteur VIII ; 4, 15-17
– préférer la pose de stent nu (BMS) plutôt que des stents actifs (DES) car les premiers nécessitent une double antiagrégation plaquettaire associant aspirine et clopidogrel pour une durée de 1 mois, relayée ensuite par l’aspirine à faible posologie, mais nous avons vu que ce point pouvait être discuté ;15-17
– éviter4 le recours à la desmopressine compte tenu des effets hémodynamiques sur les vaisseaux sanguins chez ces sujets âgés le plus souvent hypertendus ;
– effectuer un suivi rapproché du patient pour estimer les complications hémorragiques permet d’orienter la stratégie thérapeutique, soit en arrêtant le traitement antiagrégant, soit en renforçant ou en instaurant un traitement substitutif si le traitement antiagrégant doit absolument être maintenu ;
– en situation d’urgence, une anticoagulation peut être nécessaire, et les données de la littérature concernent le recours à l’héparine non fractionnée et aux héparines de bas poids moléculaire (HBPM) sans bolus sous couvert d’une substitution par facteur VIII-facteur Willebrand.4, 15-17
En cas d’angioplastie, les stents actifs (drug eluting stent [DES]) récents avec des polymères bioabsorbables permettent de diminuer la durée de la double antiagrégation plaquettaire à 1 mois comme cela est préconisé en cas de pose d’un stent nu (bare metal stent [BMS])12. Il a été démontré que les stents actifs présentent moins de risque de thrombose que les stents nus.13, 14 Ainsi, il paraîtrait intéressant d’associer les stents actifs à polymères résorbables et une double antiagrégation plaquettaire pour les patients âgés ayant une maladie de Willebrand et, dans une plus large mesure, tout patient ayant une maladie hémorragique. Néanmoins, à notre connaissance, aucune publication n’est parue à ce jour.
À l’heure actuelle, selon les données publiées, il faut pour les patients ayant une maladie de Willebrand : 4, 15-17
– évaluer précisément le phénotype hémorragique du patient par un interrogatoire exhaustif des antécédents hémorragiques ; 4, 15-17
– lors d’une coronarographie et/ou d’une angioplastie, privilégier la voie d’abord radiale car elle permet une compression du point de ponction plus facile qu’une voie d’abord fémorale et limite le risque hémorragique ; 4, 15-17
– en cas de procédures invasives, effectuer le geste sous couvert d’un traitement substitutif de concentrés de facteur Willebrand associé ou non au facteur VIII, 30 à 60 minutes avant et 24 à 48 heures après. La cible thérapeutique de la substitution est située au pic entre 80 et 100 % de VWF:RCo et du facteur VIII ; 4, 15-17
– préférer la pose de stent nu (BMS) plutôt que des stents actifs (DES) car les premiers nécessitent une double antiagrégation plaquettaire associant aspirine et clopidogrel pour une durée de 1 mois, relayée ensuite par l’aspirine à faible posologie, mais nous avons vu que ce point pouvait être discuté ;15-17
– éviter4 le recours à la desmopressine compte tenu des effets hémodynamiques sur les vaisseaux sanguins chez ces sujets âgés le plus souvent hypertendus ;
– effectuer un suivi rapproché du patient pour estimer les complications hémorragiques permet d’orienter la stratégie thérapeutique, soit en arrêtant le traitement antiagrégant, soit en renforçant ou en instaurant un traitement substitutif si le traitement antiagrégant doit absolument être maintenu ;
– en situation d’urgence, une anticoagulation peut être nécessaire, et les données de la littérature concernent le recours à l’héparine non fractionnée et aux héparines de bas poids moléculaire (HBPM) sans bolus sous couvert d’une substitution par facteur VIII-facteur Willebrand.4, 15-17
Fibrillation atriale
Le principe du traitement, pour prévenir les complications cardio-emboliques, notamment l’accident vasculaire cérébral ischémique, repose sur une anticoagulation efficace. Cette anticoagulation est indiquée chez les patients avec un score de risque CHA2DS2−VASc supérieur ou égal à 2 chez l’homme et à 3 chez la femme. Les thérapeutiques disponibles les plus fréquemment utilisées sont l’héparine non fractionnée et les HBPM, les antagonistes de la vitamine K (AVK), les anticoagulants oraux directs.
Dans l’urgence, il est toujours possible d’utiliser l’héparine non fractionnée qui a une durée de vie courte mais qui est difficile à manier et à équilibrer chez les sujets âgés, le plus souvent insuffisants rénaux. Ce traitement peut entraîner par ailleurs une thrombopénie induite par l’héparine. Les HBPM, avec leur demi-vie d’environ 4 heures, ont une réponse prédictible en fonction du poids du patient et diminuent considérablement le risque de thrombopénie. Chez les sujets âgés, il faut aussi tenir compte de l’estimation de la fonction rénale évaluée par la clairance de la créatinine calculée à l’aide de la formule de Cockcroft pour anticiper un éventuel risque d’accumulation d’activité anti-Xa et se trouver en situation de surdosage augmentant le risque d’hémorragie sévère.18 On peut surveiller, en cas de besoin, l’activité anti-Xa au pic (4 à 6 heures après l’injection) pour détecter une situation de surdosage.
Pour une anticoagulation per os, les traitements de référence sont les AVK, notamment la warfarine. L’objectif est d’obtenir un international normalized ratio (INR) entre 2 et 3. Une antagonisation de l’AVK est d’utilisation relativement simple en cas de surdosage symptomatique ou non.
Même si l’utilisation des anticoagulants oraux directs (AOD), associée à une réduction des saignements intracérébraux par rapport aux AVK, semble séduisante, aucune publication n’a porté sur les sujets atteints de maladie de Willebrand (ou de toute autre pathologie hémorragique constitutionnelle). Par ailleurs, à l’heure actuelle, seul le dabigatran dispose d’un antidote en cas de complication hémorragique. Pour le rivaroxaban et l’apixaban, le seul recours possible en cas d’hémorragie grave repose sur l’administration de concentrés de complexes prothrombiques (médicaments dérivés du sang contenant les facteurs II, VII, IX et X).
Aucune donnée ne permet d’évaluer le risque hémorragique des sujets atteints de maladie de Willebrand traités par AOD ou AVK pour une fibrillation atriale : une étude spécifique serait d’ailleurs difficile à mettre en œuvre pour obtenir un effectif de patients suffisant. Aussi, il paraît raisonnable à ce jour de traiter ces patients avec des AVK compte tenu du recul pratique dont on dispose chez les sujets non atteints de maladie de Willebrand et de contre-indiquer pour le moment l’utilisation des AOD.
À noter que des données concernant les hémophiles sont plus nombreuses. Le risque chez le sujet âgé hémophile d’avoir une fibrillation atriale est similaire à celui de la population générale âgée et, de même que chez le sujet non hémophile, il est important d’utiliser le score CHA2DS2−VASc en stratifiant l’escalade thérapeutique.19-21 Cette stratégie doit aussi laisser la place à un traitement substitutif par facteur VIII.17, 22
Il paraît possible de transposer ce raisonnement chez les patients atteints de maladie de Willebrand :
– évaluation du score CHA2DS2−VASc ;
– traitement anticoagulant décidé en fonction de ce score et prise en compte de l’insuffisance rénale pour le traitement héparinique, puis relais par AVK sous traitement substitutif prophylactique de concentrés de facteur Willebrand en fonction de l’anamnèse hémorragique du patient et de ses taux de VWF:RCo et facteur VIII. Ce traitement substitutif peut être maintenu aussi longtemps que nécessaire au cas par cas et à l’appréciation du clinicien, avec un objectif thérapeutique de VWF:RCo au moins à 30 %. Le recours à la fermeture de l’auricule gauche, en plein essor, est également une solution séduisante pour les patients avec un risque hémorragique et/ou thromboembolique élevé permettant d’éviter l’anticoagulation.17
Dans l’urgence, il est toujours possible d’utiliser l’héparine non fractionnée qui a une durée de vie courte mais qui est difficile à manier et à équilibrer chez les sujets âgés, le plus souvent insuffisants rénaux. Ce traitement peut entraîner par ailleurs une thrombopénie induite par l’héparine. Les HBPM, avec leur demi-vie d’environ 4 heures, ont une réponse prédictible en fonction du poids du patient et diminuent considérablement le risque de thrombopénie. Chez les sujets âgés, il faut aussi tenir compte de l’estimation de la fonction rénale évaluée par la clairance de la créatinine calculée à l’aide de la formule de Cockcroft pour anticiper un éventuel risque d’accumulation d’activité anti-Xa et se trouver en situation de surdosage augmentant le risque d’hémorragie sévère.18 On peut surveiller, en cas de besoin, l’activité anti-Xa au pic (4 à 6 heures après l’injection) pour détecter une situation de surdosage.
Pour une anticoagulation per os, les traitements de référence sont les AVK, notamment la warfarine. L’objectif est d’obtenir un international normalized ratio (INR) entre 2 et 3. Une antagonisation de l’AVK est d’utilisation relativement simple en cas de surdosage symptomatique ou non.
Même si l’utilisation des anticoagulants oraux directs (AOD), associée à une réduction des saignements intracérébraux par rapport aux AVK, semble séduisante, aucune publication n’a porté sur les sujets atteints de maladie de Willebrand (ou de toute autre pathologie hémorragique constitutionnelle). Par ailleurs, à l’heure actuelle, seul le dabigatran dispose d’un antidote en cas de complication hémorragique. Pour le rivaroxaban et l’apixaban, le seul recours possible en cas d’hémorragie grave repose sur l’administration de concentrés de complexes prothrombiques (médicaments dérivés du sang contenant les facteurs II, VII, IX et X).
Aucune donnée ne permet d’évaluer le risque hémorragique des sujets atteints de maladie de Willebrand traités par AOD ou AVK pour une fibrillation atriale : une étude spécifique serait d’ailleurs difficile à mettre en œuvre pour obtenir un effectif de patients suffisant. Aussi, il paraît raisonnable à ce jour de traiter ces patients avec des AVK compte tenu du recul pratique dont on dispose chez les sujets non atteints de maladie de Willebrand et de contre-indiquer pour le moment l’utilisation des AOD.
À noter que des données concernant les hémophiles sont plus nombreuses. Le risque chez le sujet âgé hémophile d’avoir une fibrillation atriale est similaire à celui de la population générale âgée et, de même que chez le sujet non hémophile, il est important d’utiliser le score CHA2DS2−VASc en stratifiant l’escalade thérapeutique.19-21 Cette stratégie doit aussi laisser la place à un traitement substitutif par facteur VIII.17, 22
Il paraît possible de transposer ce raisonnement chez les patients atteints de maladie de Willebrand :
– évaluation du score CHA2DS2−VASc ;
– traitement anticoagulant décidé en fonction de ce score et prise en compte de l’insuffisance rénale pour le traitement héparinique, puis relais par AVK sous traitement substitutif prophylactique de concentrés de facteur Willebrand en fonction de l’anamnèse hémorragique du patient et de ses taux de VWF:RCo et facteur VIII. Ce traitement substitutif peut être maintenu aussi longtemps que nécessaire au cas par cas et à l’appréciation du clinicien, avec un objectif thérapeutique de VWF:RCo au moins à 30 %. Le recours à la fermeture de l’auricule gauche, en plein essor, est également une solution séduisante pour les patients avec un risque hémorragique et/ou thromboembolique élevé permettant d’éviter l’anticoagulation.17
Événements veineux thrombotiques
Les études disponibles ne sont pas parvenues à incriminer le facteur Willebrand comme facteur de risque de la maladie thromboembolique veineuse de façon claire. Il semble qu’il n’en soit pas un facteur prédictif.23
Des patients ayant une maladie de Willebrand essentiellement de type 1 ont été comparés à une population témoin lorsqu’ils avaient une thrombose veineuse profonde et/ou une embolie pulmonaire. Le pourcentage de patients ayant une maladie de Willebrand était de 2,5 % (4/198) pour une thrombose veineuse profonde et de 1 % (2/198) pour une embolie pulmonaire sans différence significative avec le groupe de patients témoins. Finalement, il ressort de ces résultats que l’âge avancé, les comorbidités de type cancer ou les situations chirurgicales sont des facteurs de risque d’un événement thromboembolique chez tout patient, qu’il ait une maladie de Willebrand ou non.23
De même, une équipe a retenu essentiellement comme facteurs de risque thrombotique dans une cohorte de patients atteints de maladie de Willebrand : un âge supérieur à 65 ans, le recours à un traitement substitutif en facteur VIII-facteur Willebrand ou à la desmopressine, une thrombophilie et la chirurgie.24
Les recommandations internationales relatives à la prise en charge des patients atteints de maladie de Willebrand précisent bien que pour toute procédure invasive, il est important de ne pas dépasser des taux de VWF:RCo de 250 % et de facteur VIII de 250 %, associés à un risque thrombotique accru.25, 26 En cas de thrombose veineuse, la conduite à tenir est d’avoir recours aux mêmes classes thérapeutiques de traitement que celles prescrites en cas de fibrillation atriale (v. supra) : héparine non fractionnée, HBPM et AVK. Pour les traitements hépariniques chez les sujets âgés, il faut bien tenir compte de l’estimation de la fonction rénale évaluée par la clairance de la créatinine calculée à l’aide de la formule de Cockcroft et Gault pour éviter toute accumulation d’activité anti-Xa et une situation de surdosage augmentant encore plus le risque, chez ces patients, de développer une complication hémorragique sévère. Ainsi, il serait préférable de traiter les patients ayant une maladie de Willebrand avec la tinzaparine compte tenu de la tendance à moindre accumulation chez le sujet âgé.
Pour les traitements anticoagulants oraux, comme dans les fibrillations atriales, les AVK sont préférés aux AOD pour leur contrôle aisé de l’INR et la possibilité d’une antagonisation relativement simple.
Des patients ayant une maladie de Willebrand essentiellement de type 1 ont été comparés à une population témoin lorsqu’ils avaient une thrombose veineuse profonde et/ou une embolie pulmonaire. Le pourcentage de patients ayant une maladie de Willebrand était de 2,5 % (4/198) pour une thrombose veineuse profonde et de 1 % (2/198) pour une embolie pulmonaire sans différence significative avec le groupe de patients témoins. Finalement, il ressort de ces résultats que l’âge avancé, les comorbidités de type cancer ou les situations chirurgicales sont des facteurs de risque d’un événement thromboembolique chez tout patient, qu’il ait une maladie de Willebrand ou non.23
De même, une équipe a retenu essentiellement comme facteurs de risque thrombotique dans une cohorte de patients atteints de maladie de Willebrand : un âge supérieur à 65 ans, le recours à un traitement substitutif en facteur VIII-facteur Willebrand ou à la desmopressine, une thrombophilie et la chirurgie.24
Les recommandations internationales relatives à la prise en charge des patients atteints de maladie de Willebrand précisent bien que pour toute procédure invasive, il est important de ne pas dépasser des taux de VWF:RCo de 250 % et de facteur VIII de 250 %, associés à un risque thrombotique accru.25, 26 En cas de thrombose veineuse, la conduite à tenir est d’avoir recours aux mêmes classes thérapeutiques de traitement que celles prescrites en cas de fibrillation atriale (v. supra) : héparine non fractionnée, HBPM et AVK. Pour les traitements hépariniques chez les sujets âgés, il faut bien tenir compte de l’estimation de la fonction rénale évaluée par la clairance de la créatinine calculée à l’aide de la formule de Cockcroft et Gault pour éviter toute accumulation d’activité anti-Xa et une situation de surdosage augmentant encore plus le risque, chez ces patients, de développer une complication hémorragique sévère. Ainsi, il serait préférable de traiter les patients ayant une maladie de Willebrand avec la tinzaparine compte tenu de la tendance à moindre accumulation chez le sujet âgé.
Pour les traitements anticoagulants oraux, comme dans les fibrillations atriales, les AVK sont préférés aux AOD pour leur contrôle aisé de l’INR et la possibilité d’une antagonisation relativement simple.
Cancers
Il est connu depuis plusieurs décennies que les sujets âgés ont une fréquence plus élevée de cancers, mais peu de références bibliographiques spécifiques concernent des patients ayant un déficit de l’hémostase et un cancer, et encore moins ceux ayant une maladie de Willebrand.27 Les cancers les plus fréquemment observés chez les patients ayant une maladie de Willebrand sont essentiellement rapportés dans une étude :27 parmi 92 patients (âge médian au diagnostic 58 ans, 82 % avaient une tumeur solide (cancers du tractus urogénital [22 %], cancers digestifs [19 %], cancers du sein [18 %]), et 18 % avaient une hémopathie dont 54 % de lymphomes non hodgkiniens. Très peu étaient associés à une infection virale par le virus de l’hépatite C ou le virus de l’immunodéficience humaine.
En revanche, le facteur Willebrand jouerait un rôle pro-apoptotique antitumoral dans le cancer, notamment dans la réduction de l’évolution métastatique.28 Concernant les patients atteints de maladie de Willebrand, il s’agit le plus souvent de cas rapportés depuis le diagnostic jusqu’à la fin de la prise en charge du processus cancéreux (décès ou rémission et surveillance).
Globalement, ces cas rapportés nous enseignent qu’il faut prendre en charge les patients atteints d’une maladie de Willebrand ou de toute autre maladie hémorragique ayant un cancer comme le seraient les sujets non atteints.29 Les traitements, chirurgie, chimiothérapie, radiothérapie se font selon les schémas habituels, en suivant les recommandations des sociétés savantes d’oncologie.30
Néanmoins, le risque hémorragique est plus important en raison de la thrombopénie liée aux traitements de chimiothérapie et/ou radiothérapie mais aussi à cause des mucites qui fragilisent les muqueuses (tractus oto-rhino-laryngé, tractus digestif). Face à ce risque augmenté, il est proposé un traitement substitutif soit lors de la période la plus à risque (chirurgie, acmé de la mucite), soit en prophylaxie, surtout pour les formes les plus sévères à adapter au cas par cas.
Les cancers de la prostate, fréquents chez les patients âgés, sont un exemple de cancer impliquant une chirurgie à risque hémorragique. Leur traitement repose essentiellement sur de la chirurgie ou a minima une procédure invasive telle que la résection transurétrale prostatique ou la biopsie transrectale de prostate. Une équipe allemande s’est intéressée à la prise en charge péri-opératoire de patients ayant une pathologie de la coagulation et un cancer de prostate.31 Sur 37 patients étudiés (âge moyen 66 ans), 11 avaient une maladie de Willebrand modérée, les 26 autres patients étaient hémophiles A ou B. Tous ont reçu un traitement substitutif permettant une bonne hémostase pour le geste. Le temps opératoire s’avérait plus long par rapport aux patients du groupe contrôle, tout comme le temps d’hospitalisation, du fait de la période de substitution mais aussi du temps de l’irrigation et de la période d’hématurie prolongés. En revanche, aucune transfusion n’avait été nécessaire.
L’enjeu pour ces patients atteints de cancer est aussi de prévenir les complications thromboemboliques avec un traitement anticoagulant prophylactique. Un groupe d’experts a proposé des recommandations pour les patients hémophiles pour plusieurs types de cancer : il ressort la nécessité de maintenir un traitement substitutif en concentrés de facteur VIII ou IX prophylactique en fonction du type de chirurgie (risque hémorragique immédiat ou différé).
Ces recommandations pourraient s’appliquer également aux patients atteints de maladie de Willebrand en suggérant qu’il est important de discuter de façon multidisciplinaire la stratégie thérapeutique à adopter en fonction de la classification de la tumeur et de l’âge du patient qui peut avoir d’autres comorbidités. Il faut discuter du recours à des concentrés de facteur Willebrand dépourvu de facteur VIII dans ces situations car le risque de thrombose est très important (âge et cancer).22
En revanche, le facteur Willebrand jouerait un rôle pro-apoptotique antitumoral dans le cancer, notamment dans la réduction de l’évolution métastatique.28 Concernant les patients atteints de maladie de Willebrand, il s’agit le plus souvent de cas rapportés depuis le diagnostic jusqu’à la fin de la prise en charge du processus cancéreux (décès ou rémission et surveillance).
Globalement, ces cas rapportés nous enseignent qu’il faut prendre en charge les patients atteints d’une maladie de Willebrand ou de toute autre maladie hémorragique ayant un cancer comme le seraient les sujets non atteints.29 Les traitements, chirurgie, chimiothérapie, radiothérapie se font selon les schémas habituels, en suivant les recommandations des sociétés savantes d’oncologie.30
Néanmoins, le risque hémorragique est plus important en raison de la thrombopénie liée aux traitements de chimiothérapie et/ou radiothérapie mais aussi à cause des mucites qui fragilisent les muqueuses (tractus oto-rhino-laryngé, tractus digestif). Face à ce risque augmenté, il est proposé un traitement substitutif soit lors de la période la plus à risque (chirurgie, acmé de la mucite), soit en prophylaxie, surtout pour les formes les plus sévères à adapter au cas par cas.
Les cancers de la prostate, fréquents chez les patients âgés, sont un exemple de cancer impliquant une chirurgie à risque hémorragique. Leur traitement repose essentiellement sur de la chirurgie ou a minima une procédure invasive telle que la résection transurétrale prostatique ou la biopsie transrectale de prostate. Une équipe allemande s’est intéressée à la prise en charge péri-opératoire de patients ayant une pathologie de la coagulation et un cancer de prostate.31 Sur 37 patients étudiés (âge moyen 66 ans), 11 avaient une maladie de Willebrand modérée, les 26 autres patients étaient hémophiles A ou B. Tous ont reçu un traitement substitutif permettant une bonne hémostase pour le geste. Le temps opératoire s’avérait plus long par rapport aux patients du groupe contrôle, tout comme le temps d’hospitalisation, du fait de la période de substitution mais aussi du temps de l’irrigation et de la période d’hématurie prolongés. En revanche, aucune transfusion n’avait été nécessaire.
L’enjeu pour ces patients atteints de cancer est aussi de prévenir les complications thromboemboliques avec un traitement anticoagulant prophylactique. Un groupe d’experts a proposé des recommandations pour les patients hémophiles pour plusieurs types de cancer : il ressort la nécessité de maintenir un traitement substitutif en concentrés de facteur VIII ou IX prophylactique en fonction du type de chirurgie (risque hémorragique immédiat ou différé).
Ces recommandations pourraient s’appliquer également aux patients atteints de maladie de Willebrand en suggérant qu’il est important de discuter de façon multidisciplinaire la stratégie thérapeutique à adopter en fonction de la classification de la tumeur et de l’âge du patient qui peut avoir d’autres comorbidités. Il faut discuter du recours à des concentrés de facteur Willebrand dépourvu de facteur VIII dans ces situations car le risque de thrombose est très important (âge et cancer).22
Chirurgie orthopédique
Les chirurgies sont répertoriées en deux groupes, celles à risque hémorragique mineur et celles à risque hémorragique majeur.25, 26 Les sujets âgés sont les plus concernés par une chirurgie orthopédique telle que la pose d’une prothèse de hanche ou de genou dont l’indication est le plus souvent liée à l’évolution d’une arthrose. Hormis chez les patients atteints d’une maladie de Willebrand de type 3 comme chez les hémophiles, ces arthroses ne sont pas liées à l’évolution d’une hémarthrose.32 Ces chirurgies orthopédiques présentent un double enjeu. Elles sont à risque hémorragique majeur mais aussi à risque thrombogène élevé.32
Comme pour tout acte chirurgical, il ne faut envisager une chirurgie orthopédique que si elle est réellement indiquée en évaluant la balance bénéfice-risque. L’acte paraît évident en cas de fracture du col du fémur, mais discutable pour une coxarthrose surtout si le patient ne se mobilise plus.
Pour gérer le risque hémorragique, une fois de plus, la desmopressine n’est pas recommandée compte tenu de l’âge avancé des patients et du niveau de risque hémorragique (risque de tachyphylaxie).25, 26
Les recommandations internationales de substitution sont celles d’une chirurgie à risque hémorragique élevé, en ayant comme objectif initial d’obtenir un VWF:RCo et un facteur VIII autour de 100 %, puis d’avoir une résiduelle de ces deux paramètres supérieure à 50 % sans jamais dépasser les 250 % pour une durée de 7 à 10 jours.25, 26 La possibilité d’une substitution prophylactique secondaire chez des patients sévères est à discuter au cas par cas.
Une équipe a montré que 23 patients atteints de maladie de Willebrand d’âge moyen 41,8 ans (âges extrêmes 10-78 ans), opérés (32 interventions au total), ont eu des complications hémorragiques (6 patients/32).32 Quatre d’entre eux étaient alors sous HBPM prophylactique. Les deux autres avaient une reprise hémorragique au moment de la réhabilitation postopératoire (un patient maladie de Willebrand type 1 et un patient maladie de Willebrand type 2M) malgré un traitement substitutif bien mené.
Néanmoins, une prophylaxie antithrombotique par HBPM doit être envisagée dans toutes les interventions chirurgicales, notamment orthopédiques, à risque élevé de thrombose veineuse, si le risque hémorragique est maîtrisé.25, 33
Comme pour tout acte chirurgical, il ne faut envisager une chirurgie orthopédique que si elle est réellement indiquée en évaluant la balance bénéfice-risque. L’acte paraît évident en cas de fracture du col du fémur, mais discutable pour une coxarthrose surtout si le patient ne se mobilise plus.
Pour gérer le risque hémorragique, une fois de plus, la desmopressine n’est pas recommandée compte tenu de l’âge avancé des patients et du niveau de risque hémorragique (risque de tachyphylaxie).25, 26
Les recommandations internationales de substitution sont celles d’une chirurgie à risque hémorragique élevé, en ayant comme objectif initial d’obtenir un VWF:RCo et un facteur VIII autour de 100 %, puis d’avoir une résiduelle de ces deux paramètres supérieure à 50 % sans jamais dépasser les 250 % pour une durée de 7 à 10 jours.25, 26 La possibilité d’une substitution prophylactique secondaire chez des patients sévères est à discuter au cas par cas.
Une équipe a montré que 23 patients atteints de maladie de Willebrand d’âge moyen 41,8 ans (âges extrêmes 10-78 ans), opérés (32 interventions au total), ont eu des complications hémorragiques (6 patients/32).32 Quatre d’entre eux étaient alors sous HBPM prophylactique. Les deux autres avaient une reprise hémorragique au moment de la réhabilitation postopératoire (un patient maladie de Willebrand type 1 et un patient maladie de Willebrand type 2M) malgré un traitement substitutif bien mené.
Néanmoins, une prophylaxie antithrombotique par HBPM doit être envisagée dans toutes les interventions chirurgicales, notamment orthopédiques, à risque élevé de thrombose veineuse, si le risque hémorragique est maîtrisé.25, 33
Angiodysplasies digestives
Les angiodysplasies digestives sont des anomalies de développement des vaisseaux entre des capillaires de la muqueuse digestive et le réseau veineux sous-muqueux dont la fréquence augmente avec l’âge. On les retrouve chez deux tiers des sujets de plus de 70 ans et leur incidence est 200 fois plus élevée pour les patients âgés de plus de 90 ans que pour les adultes de la trentaine.34 Les angiodysplasies digestives peuvent être localisées tout le long du tube digestif, certaines études les retrouvent de façon privilégiée au niveau de l’intestin grêle, essentiellement au niveau du jéjunum et du duodénum.34 Leur physiopathologie n’est pas précisément définie, mais il y aurait un phénomène d’angiogenèse anormale.
Dans la population générale, les angiodysplasies digestives peuvent rester asymptomatiques. Le facteur Willebrand est sensible aux forces de cisaillement qui sont augmentées dans les petits capillaires des angiodysplasies, d’où le fait qu’un déficit en facteur Willebrand soit particulièrement parlant cliniquement au sein d’une angiodysplasie digestive hémorragique.35
Les publications associant anomalies du facteur Willebrand et angiodysplasies sont nombreuses et il s’agit le plus souvent de syndromes de Willebrand acquis. On trouve une association entre syndrome de Willebrand acquis et angiodysplasies digestives hémorragiques chez les sujets âgés ayant un rétrécissement aortique qui induit une perte des multimères de haut poids moléculaire du facteur Willebrand.35
Les hémorragies digestives sont plus importantes chez les sujets atteints de maladie de Willebrand congénitale, en particulier dans le type 2A et le type 3. Il existe plusieurs moyens de diagnostiquer ces angiodysplasies : la fibroscopie haute ou basse, la vidéocapsule, l’iconographie tomodensitométrique en période hémorragique. Les procédures thérapeutiques sont multiples et choisies par les spécialistes : médicamenteuses (antifibrinolytique, hormonothérapie, somatostatine)36 ou perendoscopique par coagulation au plasma argon, par électrocoagulation, par photocoagulation ou chirurgicale.36
La prise en charge chez les sujets âgés atteints de maladie de Willebrand et d’angiodysplasie digestive doit être multidisciplinaire. Le recours à la desmopressine étant le plus souvent contre-indiqué chez les sujets âgés,25, 26 la substitution est la seule option thérapeutique possible chez ces patients. Il faut à la fois prescrire un traitement substitutif initial permettant d’atteindre un VWF:RCo et/ou un facteur VIII résiduels supérieurs à 50 %,25, 26 et choisir le meilleur traitement local par voie endoscopique pour contrôler l’hémorragie. L’option chirurgicale est la dernière à être choisie. Bien souvent, il faut transfuser également des concentrés érythrocytaires. La durée du traitement substitutif doit être adaptée à l’évolution de l’événement hémorragique.25, 26 Il a été montré que la durée du traitement apparaît plus longue de 4 à 23 jours par rapport à d’autres localisations anatomiques hémorragiques.37
Il faut également discuter du recours à un traitement substitutif prophylactique par concentrés de facteur Willebrand ou de facteur Willebrand-facteur VIII si les épisodes hémorragiques sont répétés ou graves.25 Plusieurs analyses ont montré l’efficacité d’une prophylaxie au long cours pour des patients ayant des saignements gastro-intestinaux chroniques dont la cure chirurgicale est impossible. Le rythme le plus souvent décrit est d’une injection 3 fois par semaine.38
Dans la population générale, les angiodysplasies digestives peuvent rester asymptomatiques. Le facteur Willebrand est sensible aux forces de cisaillement qui sont augmentées dans les petits capillaires des angiodysplasies, d’où le fait qu’un déficit en facteur Willebrand soit particulièrement parlant cliniquement au sein d’une angiodysplasie digestive hémorragique.35
Les publications associant anomalies du facteur Willebrand et angiodysplasies sont nombreuses et il s’agit le plus souvent de syndromes de Willebrand acquis. On trouve une association entre syndrome de Willebrand acquis et angiodysplasies digestives hémorragiques chez les sujets âgés ayant un rétrécissement aortique qui induit une perte des multimères de haut poids moléculaire du facteur Willebrand.35
Les hémorragies digestives sont plus importantes chez les sujets atteints de maladie de Willebrand congénitale, en particulier dans le type 2A et le type 3. Il existe plusieurs moyens de diagnostiquer ces angiodysplasies : la fibroscopie haute ou basse, la vidéocapsule, l’iconographie tomodensitométrique en période hémorragique. Les procédures thérapeutiques sont multiples et choisies par les spécialistes : médicamenteuses (antifibrinolytique, hormonothérapie, somatostatine)36 ou perendoscopique par coagulation au plasma argon, par électrocoagulation, par photocoagulation ou chirurgicale.36
La prise en charge chez les sujets âgés atteints de maladie de Willebrand et d’angiodysplasie digestive doit être multidisciplinaire. Le recours à la desmopressine étant le plus souvent contre-indiqué chez les sujets âgés,25, 26 la substitution est la seule option thérapeutique possible chez ces patients. Il faut à la fois prescrire un traitement substitutif initial permettant d’atteindre un VWF:RCo et/ou un facteur VIII résiduels supérieurs à 50 %,25, 26 et choisir le meilleur traitement local par voie endoscopique pour contrôler l’hémorragie. L’option chirurgicale est la dernière à être choisie. Bien souvent, il faut transfuser également des concentrés érythrocytaires. La durée du traitement substitutif doit être adaptée à l’évolution de l’événement hémorragique.25, 26 Il a été montré que la durée du traitement apparaît plus longue de 4 à 23 jours par rapport à d’autres localisations anatomiques hémorragiques.37
Il faut également discuter du recours à un traitement substitutif prophylactique par concentrés de facteur Willebrand ou de facteur Willebrand-facteur VIII si les épisodes hémorragiques sont répétés ou graves.25 Plusieurs analyses ont montré l’efficacité d’une prophylaxie au long cours pour des patients ayant des saignements gastro-intestinaux chroniques dont la cure chirurgicale est impossible. Le rythme le plus souvent décrit est d’une injection 3 fois par semaine.38
LA MALADIE HÉMORRAGIQUE NE DOIT PAS LIMITER LE TRAITEMENT DES COMORBIDITÉS
L’espérance de vie des patients atteints de maladie de Willebrand tend à rejoindre celle de la population générale. Les sujets âgés atteints de la maladie de Willebrand sont donc susceptibles d’avoir les mêmes comorbidités et leurs complications que la population générale. La maladie hémorragique ne devrait pas être un facteur limitant le traitement adéquat de ces comorbidités dans les pays développés, afin d’éviter les complications, tout en en ayant soin de ne pas négliger la majoration du risque hémorragique potentiellement induit par les différentes thérapeutiques de ces morbidités. Il faut toujours réaliser les procédures invasives si et seulement si leur indication est formelle pour que cela ne soit pas une perte de chance pour le patient. Une prise en charge multidisciplinaire avec les spécialistes d’organe concernés et les hématologues référents en hémostase est indispensable.
Dans ce contexte, les recommandations nationales et internationales propres à chaque comorbidité devraient être appliquées dans la mesure du possible comme chez n’importe quel autre patient. Il faut discuter comment substituer en produits dérivés du sang le patient âgé ayant une maladie de Willebrand autant que nécessaire en s’appuyant à la fois sur les recommandations existantes et les risques propres aux comorbidités à traiter.
Dans ce contexte, les recommandations nationales et internationales propres à chaque comorbidité devraient être appliquées dans la mesure du possible comme chez n’importe quel autre patient. Il faut discuter comment substituer en produits dérivés du sang le patient âgé ayant une maladie de Willebrand autant que nécessaire en s’appuyant à la fois sur les recommandations existantes et les risques propres aux comorbidités à traiter.
Références
1. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006;4:2103-14.
2. Sanders YV, Giezenaar MA, Laros-van Gorkom BA, et al. von Willebrand disease and aging: an evolving phenotype. J Thromb Haemost 2014;12:1066-75.
3. Miesbach W, Berntorp E. When von Willebrand disease comes into age – a matter of change? Eur J Haematol 2011;86:496-501.
4. Franchini M, Coppola A. Atherothrombosis in von Willebrand disease: an analysis of the literature and implications for clinical management. Semin Thromb Hemost 2012;38:185-99.
5. Alesci RS, Krekeler S, Seifried E, Miesbach W. Do patients with haemophilia and von Willebrand disease with arterial hypertension have bleeding complications: a German single centre cohort. Blood Coagul Fibrinolysis 2012;23:320-3.
6. Seaman CD, Yabes J, Cormer DM, Ragni MV. Does deficiency of von Willebrand factor protect against cardiovascular disease? Analysis of a national discharge register. J Thromb Haemost 2015;13:1999-2003.
7. Sanders YV, Eikenboom J, de Wee EM, et al. Reduced prevalence of arterial thrombosis in von Willebrand disease. J Thromb Haemost 2013;11:845-54.
8. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013;128:e240-327.
9. Kristensen SD, Knuuti J, Saraste, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur J Anaesthesiol 2014;31:517-73.
10. Piepoli MF, Hoes AW, Agewall S, et al. 2016 European guidelines on cardiovascular disease prevention in clinical practice: the sixth joint task force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European association for Cardiovascular Prevention and Rehabilitation (EACPR). Eur Heart J 2016;37:2315-81.
11. Li L, Geraghty OC, Mehta Z, Rothwell PM, Oxford Vascular Study. Age-specific risks, severity, time course, and outcome of bleeding on long-term antiplatelet treatment after vascular events: a population-based cohort study. Lancet 2017;390:490-9.
12. Urban P, Meredith IT, Abizaid A, et al. Polymer-free drug-coated coronary stents in patients at high bleeding risk. N Engl J Med 2015;373:2038-47.
13. Morice MC, Talwar S, Gaemperli O, et al. Drug-coated versus bare metal stents for elderly patients: a predefined sub-study of the LEADERS FREE trial. Int J Cardiol 2017;243:110-5.
14. Varenne O, Cook S, Sideris G, et al. Drug-eluting stents in elderly patients with coronary artery disease (SENIOR): a randomized single-blind trial. Lancet 2018;391:41-50.
15. Hassan SA, Amer S, Qureshi W, Alirhayim Z, Kuriakose P. Treating symptomatic coronary artery disease in patients with von Willebrand disease. Hematol Oncol Stem Cell Ther 2013;6:101-4.
16. Lim MY, Pruthi RK. Outcomes of management of acute coronary syndrome in patients with congenital bleeding disorders: a single center experience and review of the literature. Thromb Res 2012;130:316-22.
17. Martin K, Key NS. How I treat patients with inherited bleeding disorders who need anticoagulant therapy? Blood 2016;128:178-84.
18. Siguret V, Gouin-Thibault I, Gaussem P, Pautas E. Optimizing the use of anticoagulants (heparins and oral anticoagulants) in the elderly. Drugs Aging 2013;30:687-99.
19. Cayla G, Morange P, Chambost H, Schved JF. Management of cardiovascular disease in haemophilia. Thromb Res 2013;132:8-14.
20. Zimmermann R, Staritz P, Huth-Kühne A. Challenges in treating elderly patients with haemophilia: a focus on cardiology. Thromb Res 2014;134:S48-52.
21. Schutgens RE, Klamroth R, Pabinger I, Malerba M, Dolan G, ADVANCE Working Group. Atrial fibrillation in patients with haemophilia: a cross-sectional evaluation in Europe. Haemophilia 2014;20:682-6.
22. Mannucci PM, Schutgens RE, Santagostino E, Mauser-Bunschoten EP. How I treat age-related morbidities in elderly persons with hemophilia? Blood 2009;114:5256-63.
23. Qureshi W, Hassan S, Dabak V, Kuriakose P. Thrombosis in von Willebrand disease. Thromb Res 2012;130:e255-8.
24. Girolami A, Tasinato V, Sambado L, Peroni E, Casonato A. Venous thrombosis in von Willebrand disease as observed in one center and as reported in the literature. Blood Coagul Fibrinolysis 2015;26:54-8.
25. Mannucci PM, Franchini M, Castaman G, Federici AB. Italian Association of Hemophilia Centers. Evidence-based recommendations on the treatment of von Willebrand disease in Italy. Blood Transfus 2009;7:117-26.
26. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD: evidence-based diagnosis and management guideline, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008;14:171-232.
27. Franchini M, Di Perna C, Santoro C, et al. Italian Association of Haemophilia Centres. Cancers in patients with von Willebrand disease: A Survey from the Italian Association of Haemophilia Centres. Semin Thromb Hemost 2016;42:36-41.
28. Starke RD, Ferraro F, Paschalaki KE, Mann GE, Hunter MG, Robinson RS. Endothelial von Willebrand factor regulates angiogenesis. Blood 2011;117:1071-80.
29. Gallego L, Junquera L. Association von Willebrand’s disease and nevoid basal cell carcinoma syndrome (Gorlin syndrome). Haemophilia 2008;14:835-7.
30. Biron-Andreani C, de Moerloose P, D’Oiron R, Chambost H, Schved JF, Hermans C. Cancer detection and management in patients with haemophilia: a retrospective European multicentre study. Haemophilia 2014;20:78-82.
31. Rogenhafer S, Hauser S, Breuer A, Fechner G, Mueller SC, Oldenburg J, Goldmann G. Urological surgery in patients with hemorrhagic bleeding disorders Hemophilia A, Hemophilia B, von Willebrand disease: a retrospective study with matched pairs analysis. World J Urol 2013;31:703-7.
32. Siboni SM, Biguzzi E, Solimeno LP, et al. Orthopaedic surgery in patients with von Willebrand Disease. Haemophilia 2014;20:133-40.
33. Franchini M, Targher G, Montagnana M, Lippi G. Antithrombotic prophylaxis in patients with von Willebrand disease undergoing major surgery: when is it necessary? J Thromb Thrombolysis 2009;28:215-9.
34. Compagna R, Serra R, Sivero L, et al. Tailored treatment of intestinal angiodysplasia in elderly. Open Med (Wars) 2015;10:538-42.
35. Veyradier A, Balian A, Wolf M, et al. Abnormal von Willebrand factor in bleeding angiodysplasias of the digestive tract. Gastroenterology 2001;20:346-53.
36. Beg S, Ragunath K. Review on gastrointestinal angiodysplasia throughout the gastrointestinal tract. Pract Res Clin Gastroenterol 2017;31:119-25.
37. Berntorp E, Windyga J; European Wilate study group. Treatment and prevention of acute bleedings in von Willebrand disease-efficacy and safety of Wilate, a new generation von Willebrand factor/factor VIII concentrate. Haemophilia 2009;15:122-30.
38. Franchini M, Mannucci PM. Von Willebrand disease-associated angiodysplasia: a few answers, still many questions. Br J Haematol 2013;161:177-82.
2. Sanders YV, Giezenaar MA, Laros-van Gorkom BA, et al. von Willebrand disease and aging: an evolving phenotype. J Thromb Haemost 2014;12:1066-75.
3. Miesbach W, Berntorp E. When von Willebrand disease comes into age – a matter of change? Eur J Haematol 2011;86:496-501.
4. Franchini M, Coppola A. Atherothrombosis in von Willebrand disease: an analysis of the literature and implications for clinical management. Semin Thromb Hemost 2012;38:185-99.
5. Alesci RS, Krekeler S, Seifried E, Miesbach W. Do patients with haemophilia and von Willebrand disease with arterial hypertension have bleeding complications: a German single centre cohort. Blood Coagul Fibrinolysis 2012;23:320-3.
6. Seaman CD, Yabes J, Cormer DM, Ragni MV. Does deficiency of von Willebrand factor protect against cardiovascular disease? Analysis of a national discharge register. J Thromb Haemost 2015;13:1999-2003.
7. Sanders YV, Eikenboom J, de Wee EM, et al. Reduced prevalence of arterial thrombosis in von Willebrand disease. J Thromb Haemost 2013;11:845-54.
8. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013;128:e240-327.
9. Kristensen SD, Knuuti J, Saraste, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur J Anaesthesiol 2014;31:517-73.
10. Piepoli MF, Hoes AW, Agewall S, et al. 2016 European guidelines on cardiovascular disease prevention in clinical practice: the sixth joint task force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European association for Cardiovascular Prevention and Rehabilitation (EACPR). Eur Heart J 2016;37:2315-81.
11. Li L, Geraghty OC, Mehta Z, Rothwell PM, Oxford Vascular Study. Age-specific risks, severity, time course, and outcome of bleeding on long-term antiplatelet treatment after vascular events: a population-based cohort study. Lancet 2017;390:490-9.
12. Urban P, Meredith IT, Abizaid A, et al. Polymer-free drug-coated coronary stents in patients at high bleeding risk. N Engl J Med 2015;373:2038-47.
13. Morice MC, Talwar S, Gaemperli O, et al. Drug-coated versus bare metal stents for elderly patients: a predefined sub-study of the LEADERS FREE trial. Int J Cardiol 2017;243:110-5.
14. Varenne O, Cook S, Sideris G, et al. Drug-eluting stents in elderly patients with coronary artery disease (SENIOR): a randomized single-blind trial. Lancet 2018;391:41-50.
15. Hassan SA, Amer S, Qureshi W, Alirhayim Z, Kuriakose P. Treating symptomatic coronary artery disease in patients with von Willebrand disease. Hematol Oncol Stem Cell Ther 2013;6:101-4.
16. Lim MY, Pruthi RK. Outcomes of management of acute coronary syndrome in patients with congenital bleeding disorders: a single center experience and review of the literature. Thromb Res 2012;130:316-22.
17. Martin K, Key NS. How I treat patients with inherited bleeding disorders who need anticoagulant therapy? Blood 2016;128:178-84.
18. Siguret V, Gouin-Thibault I, Gaussem P, Pautas E. Optimizing the use of anticoagulants (heparins and oral anticoagulants) in the elderly. Drugs Aging 2013;30:687-99.
19. Cayla G, Morange P, Chambost H, Schved JF. Management of cardiovascular disease in haemophilia. Thromb Res 2013;132:8-14.
20. Zimmermann R, Staritz P, Huth-Kühne A. Challenges in treating elderly patients with haemophilia: a focus on cardiology. Thromb Res 2014;134:S48-52.
21. Schutgens RE, Klamroth R, Pabinger I, Malerba M, Dolan G, ADVANCE Working Group. Atrial fibrillation in patients with haemophilia: a cross-sectional evaluation in Europe. Haemophilia 2014;20:682-6.
22. Mannucci PM, Schutgens RE, Santagostino E, Mauser-Bunschoten EP. How I treat age-related morbidities in elderly persons with hemophilia? Blood 2009;114:5256-63.
23. Qureshi W, Hassan S, Dabak V, Kuriakose P. Thrombosis in von Willebrand disease. Thromb Res 2012;130:e255-8.
24. Girolami A, Tasinato V, Sambado L, Peroni E, Casonato A. Venous thrombosis in von Willebrand disease as observed in one center and as reported in the literature. Blood Coagul Fibrinolysis 2015;26:54-8.
25. Mannucci PM, Franchini M, Castaman G, Federici AB. Italian Association of Hemophilia Centers. Evidence-based recommendations on the treatment of von Willebrand disease in Italy. Blood Transfus 2009;7:117-26.
26. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD: evidence-based diagnosis and management guideline, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008;14:171-232.
27. Franchini M, Di Perna C, Santoro C, et al. Italian Association of Haemophilia Centres. Cancers in patients with von Willebrand disease: A Survey from the Italian Association of Haemophilia Centres. Semin Thromb Hemost 2016;42:36-41.
28. Starke RD, Ferraro F, Paschalaki KE, Mann GE, Hunter MG, Robinson RS. Endothelial von Willebrand factor regulates angiogenesis. Blood 2011;117:1071-80.
29. Gallego L, Junquera L. Association von Willebrand’s disease and nevoid basal cell carcinoma syndrome (Gorlin syndrome). Haemophilia 2008;14:835-7.
30. Biron-Andreani C, de Moerloose P, D’Oiron R, Chambost H, Schved JF, Hermans C. Cancer detection and management in patients with haemophilia: a retrospective European multicentre study. Haemophilia 2014;20:78-82.
31. Rogenhafer S, Hauser S, Breuer A, Fechner G, Mueller SC, Oldenburg J, Goldmann G. Urological surgery in patients with hemorrhagic bleeding disorders Hemophilia A, Hemophilia B, von Willebrand disease: a retrospective study with matched pairs analysis. World J Urol 2013;31:703-7.
32. Siboni SM, Biguzzi E, Solimeno LP, et al. Orthopaedic surgery in patients with von Willebrand Disease. Haemophilia 2014;20:133-40.
33. Franchini M, Targher G, Montagnana M, Lippi G. Antithrombotic prophylaxis in patients with von Willebrand disease undergoing major surgery: when is it necessary? J Thromb Thrombolysis 2009;28:215-9.
34. Compagna R, Serra R, Sivero L, et al. Tailored treatment of intestinal angiodysplasia in elderly. Open Med (Wars) 2015;10:538-42.
35. Veyradier A, Balian A, Wolf M, et al. Abnormal von Willebrand factor in bleeding angiodysplasias of the digestive tract. Gastroenterology 2001;20:346-53.
36. Beg S, Ragunath K. Review on gastrointestinal angiodysplasia throughout the gastrointestinal tract. Pract Res Clin Gastroenterol 2017;31:119-25.
37. Berntorp E, Windyga J; European Wilate study group. Treatment and prevention of acute bleedings in von Willebrand disease-efficacy and safety of Wilate, a new generation von Willebrand factor/factor VIII concentrate. Haemophilia 2009;15:122-30.
38. Franchini M, Mannucci PM. Von Willebrand disease-associated angiodysplasia: a few answers, still many questions. Br J Haematol 2013;161:177-82.
Dans cet article

Résumé
La maladie de Willebrand est une maladie hémorragique rare héréditaire (prévalence des formes symptomatiques : 1/10 000). Le facteur Willebrand augmente physiologiquement avec l’âge, d’où une diminution de la fréquence et de la sévérité de la symptomatologie hémorragique. La prise en charge des comorbidités des patients âgés doit rester multidisciplinaire et se faire au cas par cas, en adaptant les recommandations des sociétés savantes et des spécialistes de l’hémostase. Le risque hémorragique est à évaluer avant toute procédure invasive ou tout traitement pouvant majorer ce risque (anticoagulants, antiagrégants plaquettaires, certaines chimiothérapies anticancéreuses).