Le vWF est synthétisé par les cellules endothéliales et les mégacaryocytes. Il intervient dans l’hémostase primaire et permet l’adhésion des plaquettes au sous-endothélium. Il a également pour rôle de transporter le facteur VIII et de le protéger contre la dégradation.1
Trois types de maladie sont décrits : – le type 1 correspond à un déficit en vWF quantitatif et partiel (60 à 80 % des cas) ;
– le 2 à un défaut qualitatif (fonction altérée, 20 à 35 %) ;
– le 3 à un déficit quasi-total (forme la plus sévère mais extrêmement rare, avec une prévalence de 1/1 000 000).
La transmission est autosomique dominante, sauf pour les types 3 et 2N, qui sont récessifs.
On distingue les formes vraies (taux < 30 %) des déficits « low level », compris entre 30 et 50 %.
Clinique
Dans les formes symptomatiques les plus fréquentes, le tableau est dominé par des saignements muqueux (épistaxis, gingivorragies) et cutanés (ecchymoses).
En cas de maladie de type 3, en raison d’un taux de FVIII très bas (souvent< 10 %), les signes hémorragiques sont proches de ceux des hémophiles sévères : hémarthroses et arthropathies chroniques, hématomes musculaires, parfois graves.
Quand évoquer la maladie ?
Notons que, selon la Société française d’anesthésie et réanimation (SFAR), il n’est pas recommandé de réaliser un bilan d’hémostase systématique en préopératoire. En revanche, un questionnaire standardisé est indispensable pour dépister un risque hémorragique augmenté.2
En cas de symptomatologie hémorragique, le patient doit être adressé en consultation d’hémostase spécialisée pour effectuer les tests adaptés.
À l’interrogatoire, on évalue les manifestations hémorragiques. Des questionnaires sont utiles pour dépister une maladie de Willebrand, comme le score de Tosetto3 (élevé si > 4 chez l’homme et 5 chez la femme) qui prend en compte les épistaxis, les ecchymoses, les saignements prolongés pour des blessures mineures, les hémorragies digestives, du système nerveux central, les saignements lors d’extractions dentaires, au cours des procédures chirurgicales, les hématomes intramusculaires, les hémarthroses, et chez les femmes, les ménorragies et les hémorragies du post- partum.
Diagnostic : biologique
Les tests de coagulation de première intention – temps de Quick, le plus souvent exprimé en taux de prothrombine (TP), et temps de céphaline avec activateur (TCA) – ne sont pas toujours concluants. Le TCA est allongé en cas de déficit en FVIII, mais ce dernier n’est pas systématique dans la maladie de Willebrand. Ainsi, des valeurs normales n’excluent pas le diagnostic. Ce dernier repose sur le dosage de l’activité du facteur Willebrand (cofacteur de la ristocétine, vWF RCo) et de son taux circulant (vW FAg). Sont utiles : le temps d’occlusion plaquettaire mesuré par le PFA (Platelet Function Analyzer), le dosage du FVIII, l’agrégation plaquettaire à la ristocétine (RIPA) et la numération plaquettaire (
L’interprétation des résultats est délicate. Le vWF est élevé chez le nouveau-né à terme et le prématuré (> 80 %), puis diminue progressivement jusqu’à l’âge de 6-12 mois. Chez l’adulte, les valeurs de référence tant pour l’antigène que pour l’activité sont variables et comprises entre 50 et 200 %.
Le taux de vWF dépend fortement du groupe sanguin. Il est physiologiquement plus bas chez les personnes de groupe O que dans le reste de la population. Ainsi, on considère généralement qu’un taux abaissé jusqu’à 40 % est normal chez ces patients.
Par ailleurs, les taux de vWF et de FVIII augmentent en cas de syndrome inflammatoire (évalué par le dosage du fibrinogène).
Autres circonstances où le vWF est élevé : grossesse, avancée en âge, origine ethnique (Africains) ou encore stress, effort physique, néoplasie.
Dans tous les cas, si la maladie est suspectée, il convient d’adresser le patient en consultation spécialisée pour confirmer le diagnostic, établir une carte à porter 24 h/24 et la conduite à tenir en cas d’accident hémorragique ou de chirurgie. Les patients avec des formes sévères font l’objet d’un suivi spécialisé régulier.
Quelle prise en charge ?
Le PFA (Platelet Function Analyzer) a une très bonne sensibilité mais une mauvaise spécificité. S’il est anormal avec des temps d’occlusion allongés, numération plaquettaire et hématocrite normaux, on évoque une maladie de Willebrand, une thrombopathie sévère ou la prise récente de médicaments antiplaquettaires.
Les résultats des dosages de Willebrand sont interprétés en fonction du groupe sanguin et de la présence ou non d’un syndrome inflammatoire. En cas de diminution parallèle de l’antigène et de l’activité Willebrand avec un ratio RCo/Ag > 0,6-0,7, on s’oriente vers une maladie de Willebrand de type 1. Si l’activité est réduite de façon plus importante que l’antigène (RCo/Ag ≤ 0,6-0,7), on évoque un type 2. En cas de vWF indosable aussi bien en activité qu’en antigène avec un facteur VIII < 10 %, un type 3 est probable, mais le diagnostic de certitude nécessite des techniques de dosage très sensibles.
Les autres examens (étude de l’agrégation plaquettaire à la ristocétine, des multimères du facteur de Willebrand, de la capacité de liaison du vWF au FVIII, test génétique…) permettent de caractériser précisément un type 2.
Une diminution isolée du facteur VIII fait évoquer soit une hémophilie A mineure, soit une maladie de Willebrand de type 2N.
En cas de score hémorragique élevé avec des taux de vWF normaux, après répétition du bilan de Willebrand, il faut rechercher d’autres maladies hémorragiques (thrombopathies, déficits rares en facteur de la coagulation…). Le patient doit être adressé impérativement dans un centre dédié pour des analyses spécialisées (cytométrie de flux plaquettaire et agrégation plaquettaire par exemple).
Suspecter une maladie de Willebrand devant des ecchymoses, des épistaxis, des règles abondantes, des saignements prolongés pour des blessures mineures.
Le bilan standard (NFS, TP, TCA) manque de sensibilité pour le dépistage.
Le diagnostic repose sur les dosages de l’antigène et de l’activité Willebrand et du facteur VIII.
Bilan d’hémostase : quand évoquer un Willebrand ?
Le PFA (Platelet Function Analyzer) a une très bonne sensibilité mais une mauvaise spécificité. S’il est anormal avec des temps d’occlusion allongés, numération plaquettaire et hématocrite normaux, on évoque une maladie de Willebrand, une thrombopathie sévère ou la prise récente de médicaments antiplaquettaires.
Les résultats des dosages de Willebrand sont interprétés en fonction du groupe sanguin et de la présence ou non d’un syndrome inflammatoire. En cas de diminution parallèle de l’antigène et de l’activité Willebrand avec un ratio RCo/Ag > 0,6-0,7, on s’oriente vers une maladie de Willebrand de type 1. Si l’activité est réduite de façon plus importante que l’antigène (RCo/Ag ≤ 0,6-0,7), on évoque un type 2. En cas de vWF indosable aussi bien en activité qu’en antigène avec un facteur VIII < 10 %, un type 3 est probable, mais le diagnostic de certitude nécessite des techniques de dosage très sensibles.
Les autres examens (étude de l’agrégation plaquettaire à la ristocétine, des multimères du facteur de Willebrand, de la capacité de liaison du vWF au FVIII, test génétique…) permettent de caractériser précisément un type 2.
Une diminution isolée du facteur VIII fait évoquer soit une hémophilie A mineure, soit une maladie de Willebrand de type 2N.
En cas de score hémorragique élevé avec des taux de vWF normaux, après répétition du bilan de Willebrand, il faut rechercher d’autres maladies hémorragiques (thrombopathies, déficits rares en facteur de la coagulation…). Le patient doit être adressé impérativement dans un centre dédié pour des analyses spécialisées (cytométrie de flux plaquettaire et agrégation plaquettaire par exemple).
2. Bonhomme F, Ajzenberg N, Schved J, et al. Pre-interventional haemostatic assessment: Guidelines from the French Society of Anaesthesia and Intensive Care. Eur J Anaesthesiol 2013;30:142-62.
3. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost 2006;4:766-73.
4. Roberts JC, Flood VH. Laboratory diagnosis of von Willebrand disease. Int J Lab Hematol 2015;37(suppl 1):11-17.
5. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006;4:2103-114.
 Encadrés
Encadrés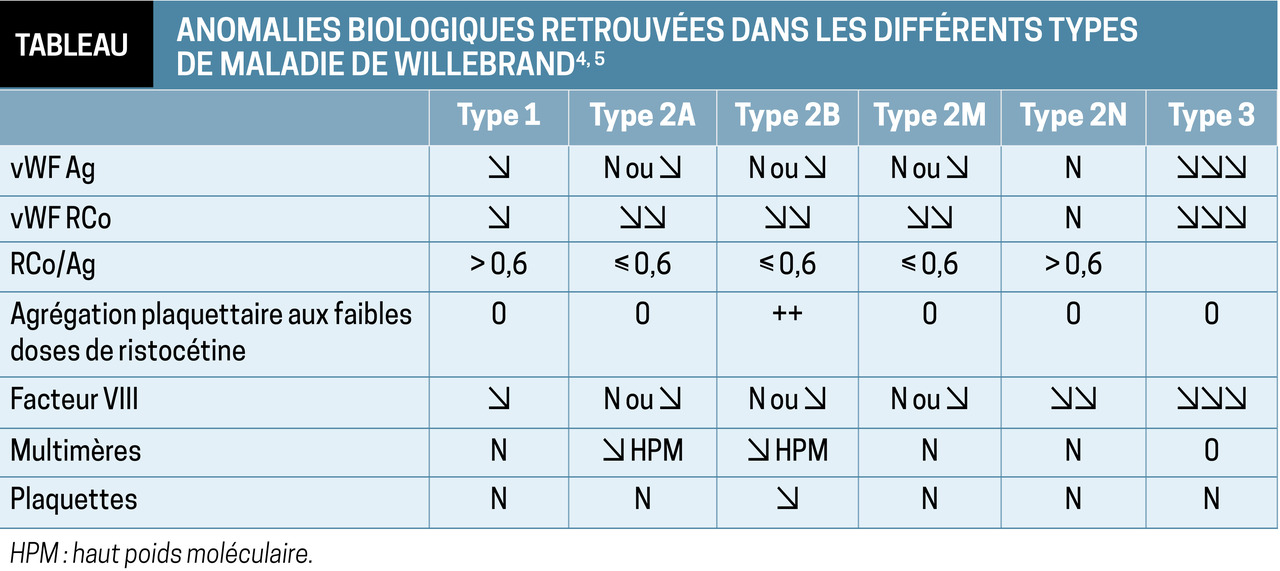
Suspecter une maladie de Willebrand devant des ecchymoses, des épistaxis, des règles abondantes, des saignements prolongés pour des blessures mineures.
Le bilan standard (NFS, TP, TCA) manque de sensibilité pour le dépistage.
Le diagnostic repose sur les dosages de l’antigène et de l’activité Willebrand et du facteur VIII.