Connaître la définition et l’organisation de prise en charge des maladies rares en France.
Connaître les principaux points d’appel devant faire évoquer une maladie rare et les circuits proposés de prise en charge.
Comprendre la notion d’errance diagnostique, savoir comment la diminuer et la distinguer de la notion d’impasse diagnostique conformément aux instructions du PNMR3.
Définitions
Maladie rare (rang A [2C-022-DE-A01])
Errance diagnostique
Impasse diagnostique
Connaître l’organisation des soins des maladies rares en France (rang B [2C-022-DE-B01])
Conformément à la vision du Consortium international pour la recherche sur les maladies rares (IRDiRC, International Rare Diseases Research Consortium), ce plan vise à ce que toutes les personnes souffrant de maladies rares aient reçu un diagnostic précis un an après la première consultation médicale spécialisée et puissent bénéficier des soins et thérapies disponibles. Les seules exceptions concernent les malades pour lesquels l’état de l’art scientifique et technique ne permet pas d’aboutir à un diagnostic précis.
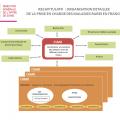
Organisation
Un centre de compétences assure la prise en charge et le suivi des personnes atteintes de maladies rares au plus proche de leur domicile, sur la base d’un maillage territorial adapté et en lien avec le CRMR dont il dépend fonctionnellement.
Une filière de santé maladies rares est une organisation qui coordonne en réseau un ensemble associant des centres de soins hospitaliers, des laboratoires de diagnostic et de recherche, des structures sociales et médico-sociales, des universités, des associations, voire tout autre partenaire ou institution apportant une expertise complémentaire au champ des maladies rares concerné (
Ressources
- 23 filières nationales de santé maladies rares en miroir des 24 réseaux européens de référence (ERN), dont 20 concernent les maladies rares ;
- 109 centres de référence maladies rares multisites formés de 387 centres de référence et de plus de 1 800 centres de compétences ou de ressources et de compétences ;
- plus de 220 associations de patients fédérées par l’Alliance maladies rares
Étiologie (rang B [2C-022-ET-B01])
L’item 45 aborde la trisomie 21, le syndrome de l’X fragile et la mucoviscidose.
Dans cet item « Maladies rares », nous évoquerons les maladies rares qui seront éventuellement rencontrées dans la carrière d’un médecin généraliste, en particulier l’hémochromatose, l’hémophilie, la drépanocytose, les thalassémies, la dystrophie musculaire de Duchenne. L’objectif n’est pas de proposer un long développement sur toutes ces maladies, mais d’appréhender leur fréquence, l’immense diversité des portes d’entrée cliniques et le nécessaire suivi pluridisciplinaire des patients.
Hémochromatose
Hémophilie
Drépanocytose
Thalassémies
Dystrophie musculaire de Duchenne
Épidémiologie (rang B [2C-022-PE-B01])
- 7 000 maladies rares différentes ;
- 80 % des maladies rares sont d’origine génétique ;
- 50 millions de patients atteints de maladies rares dans le monde ;
- 75 % des malades sont des enfants ;
- 50 % des patients n’ont pas de diagnostic précis.
- Orphanet, plateforme d’information sur les maladies rares et les médicaments orphelins en accès libre https://www.orpha.net/ ;
- Maladies rares info services, service d’information et de soutien de référence sur les maladies rares. Pour être écouté, s’informer, témoigner, échanger. https://www.maladiesraresinfo.org/ ou par téléphone : 01 56 53 81 36 ;
- les plateformes d’expertise maladies rares mises en place dans les établissements de santé abritant des CRMR et CCMR (centres de compétence maladies rares). L’objectif est d’améliorer la visibilité du parcours de soins du patient : partage d’expertise, mise en place de formations, actions médicosociales, mutualisation des connaissances et des compétences, rôle de coordination à l’échelle d’un territoire.
Prise en charge des maladies rares en ville (rang B [2C-022-PC-B01])
Conclusion
POINTS FORTS À RETENIR
Une maladie rare est une maladie qui atteint moins de 1 personne sur 2 000 en population générale.
Plus de 3 millions de personnes en France sont atteintes d’une maladie rare.
Devant des signes cliniques inhabituels et/ou un ensemble incohérent de symptômes, évoquer une maladie rare doit devenir un réflexe pour les professionnels de santé.
Chaque médecin doit connaître l’organisation nationale très structurée constituée par les centres de référence maladies rares (CRMR) et par les filières de santé maladies rares (FSMR).
L’hémochromatose, l’hémophilie, la drépanocytose, les thalassémies et la dystrophie musculaire de Duchenne font partie des maladies rares.
Les patients doivent être informés de l’existence d’associations spécifiques ou de l’Alliance maladies rares qui les orientera.
Message de l'auteur
Une Maladie rare ? Savoir y penser et trouver les informations.
Avoir la « culture du doute » devant toute association inhabituelle de symptômes ou tableau clinique atypique.
Connaitre :
– Orphanet
– Maladies rares infos service
– Alliance maladies rares
– Équipes relais handicap rares, la MDPH
– Protocoles nationaux de diagnostic et de soins (PNDS) publiés sur le site de la HAS
 Encadrés
Encadrés