Les mouvements anormaux de l’enfant ont des causes très variées. Les plus fréquents sont, d’une part, les tics et les stéréotypies et, d’autre part, les dystonies. Leur retentissement sur la vie quotidienne et la scolarité peut être majeur.
Les mouvements anormaux de l’enfant constituent un groupe très varié sur le plan phénoménologique. La démarche diagnostique repose sur un interrogatoire minutieux et une fine analyse sémiologique qui va permettre la classification du mouvement anormal. L’examen d’un jeune patient nécessite de s’adapter à son âge, pour un examen de qualité. Les vidéos familiales sont très aidantes pour le diagnostic. La caractérisation du mouvement anormal et le raisonnement anatomoclinique impliquent la prise en compte des aspects développementaux : la motricité va évoluer de la naissance à l’âge adulte, au gré de la maturation du système nerveux. Ainsi, certains mouvements peuvent être physiologiques à un moment de la vie, et pathologiques au-delà d’un certain âge : c’est le cas des syncinésies, qui disparaissent après l’âge de 6 ans en l’absence de trouble. Un mouvement anormal peut résulter du dysfonctionnement d’une des multiples structures neurologiques impliquées de la programmation à l’exécution du geste. La présentation pédiatrique de certaines maladies peut différer de ce qui est observé chez l’adulte : ainsi, une carence dopaminergique se traduit typiquement par un syndrome akinéto-rigide chez l’adulte, et par une dystonie chez l’enfant. Les mouvements anormaux de l’enfant sont plus souvent mixtes, combinant différents désordres moteurs élémentaires, tels que dystonie et chorée ou encore ataxie et spasticité.
Motif de consultation, démarche diagnostique
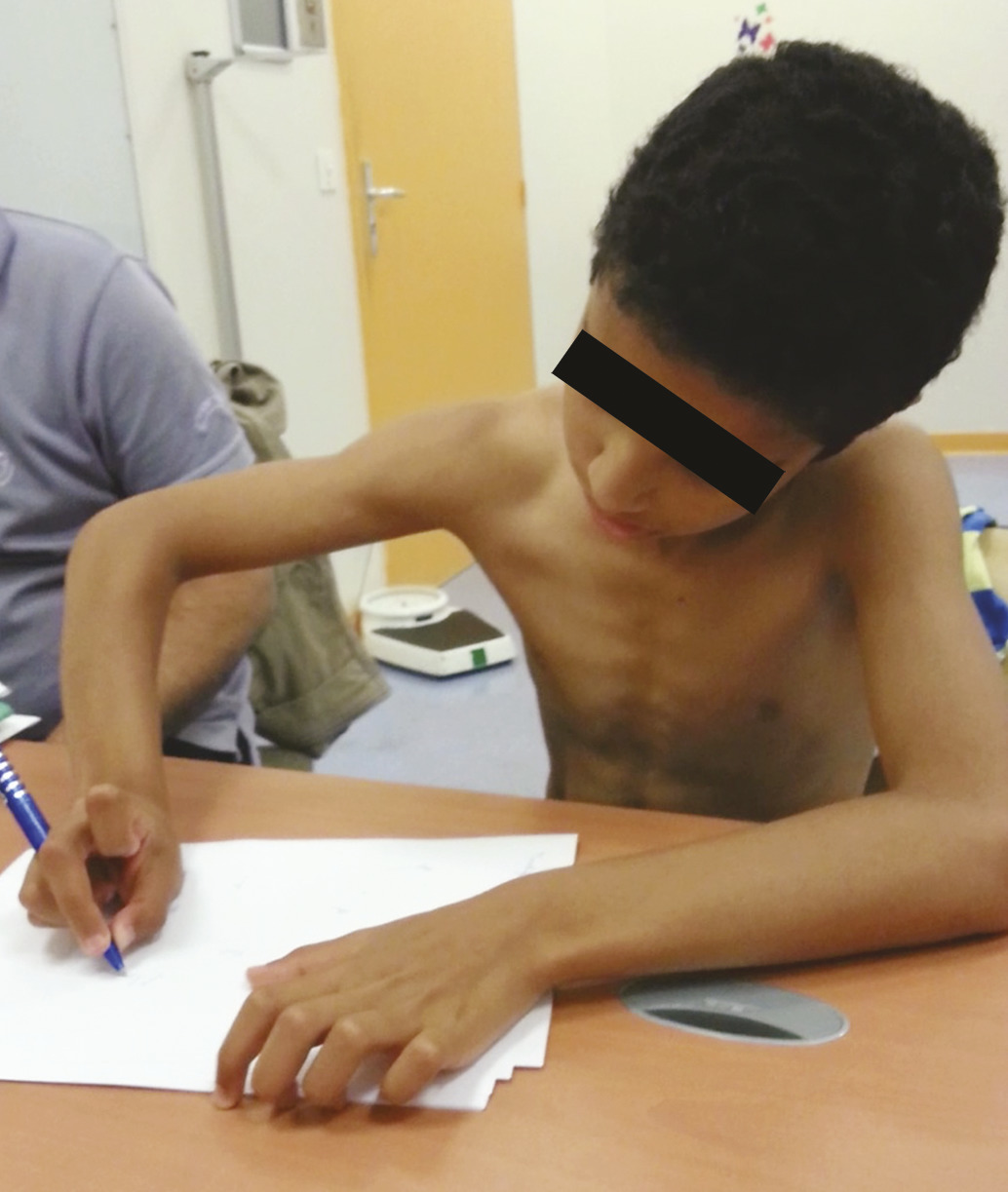
L’enfant consulte parfois pour un mouvement qui l’inquiète ou inquiète l’entourage, ou du fait du retentissement du mouvement (gêne à l’écriture, au sport, moqueries des pairs…). Les mouvements anormaux les plus fréquemment vus en consultation sont, d’une part, les troubles moteurs neurodéveloppementaux (tics et stéréotypies), d’autre part, les dystonies (fig. 1 et 2 ).
Une démarche diagnostique est proposée dans letableau 1 . Certaines causes traitables à ne pas rater sont résumées dans le tableau 2 . Les mouvements anormaux sont un motif fréquent de consultation, mais ils s’inscrivent le plus souvent dans un cadre qui permet de rassurer les familles.
Une démarche diagnostique est proposée dans le
Mouvements anormaux permanents
Chorée
La chorée est définie par des mouvements involontaires irréguliers rapides sur un fond hypotonique. Chez l’enfant, elle peut être secondaire à des lésions cérébrales (lésions anoxo-ischémiques périnatales, ou plus rarement vasculaires, tumorales…) ou à des mécanismes auto-immuns (chorée aiguë post-streptococcique de Sydenham, encéphalites à anticorps antirécepteurs N-méthyl-D-aspartate [NMDA]…). Elle peut être le symptôme prédominant d’une maladie neurodégénérative, d’où l’importance de suivre attentivement l’évolution des enfants ayant des mouvements anormaux, en étant vigilant quant à la survenue de nouveaux signes cliniques. Il existe une forme de chorée chronique avec décalage posturo-moteur, sans survenue d’autres signes neurologiques, dite chorée bénigne familiale, souvent liée à une mutation NKX2-1.1 Le traitement de la chorée dépend de la cause de celle-ci (traitements immunomodulateurs discutés dans les causes auto-immunes notamment). Les médicaments pouvant apporter un bénéfice symptomatique sont principalement la tétrabénazine, la L-dopa et le valproate de sodium.
Dystonie
La dystonie, en tant que symptôme, est définie comme une contraction musculaire tonique involontaire et soutenue, entraînant des mouvements et des postures anormales (fig. 2 ). On l’évalue cliniquement en variant les schémas moteurs (marche en arrière, course) et en testant la motricité volontaire (faire attraper un jouet…). Chez l’enfant, elle touche volontiers plusieurs parties du corps, et elle est le plus souvent secondaire. La cause principale de dystonies de l’enfant est, en effet, l’infirmité motrice cérébrale (lésions anoxo-ischémiques, ictère nucléaire néonatal). La dystonie peut aussi refléter des maladies neurométaboliques (maladie de Lesch-Nyhan, maladies mitochondriales, lysosomales, acidurie organique…). Dans tous ces cas, les mouvements anormaux sont souvent sévères et accompagnés d’autres signes neurologiques (spasticité, ataxie, atteinte cognitive) et /ou extra-neurologiques, qui vont guider l’orientation diagnostique. L’aggravation des symptômes, la dégradation cognitive et/ou motrice sont des signes d’alerte devant amener à une consultation spécialisée dans un délai rapide. L’imagerie par résonance magnétique (IRM) cérébrale est l’examen complémentaire de première intention, qui guide les analyses ultérieures. Un diagnostic d’encéphalopathie anoxo-ischémique ne doit pas être porté trop hâtivement et repose sur des critères anamnestiques et radiologiques rigoureux. En fonction de la clinique et de l’imagerie, les investigations sur le plan métabolique et génétique sont prescrites, en veillant toujours à rechercher prioritairement les causes potentiellement traitables. Les avancées génétiques récentes permettent l’identification d’un nombre croissant de syndromes dystoniques à début précoce. La place et l’accessibilité du séquençage de nouvelle génération sont en évolution mais deviendront essentielles dans les années à venir. Le diagnostic causal est un enjeu important tant pour définir les prises en charge que pour apporter aux familles des explications et un conseil génétique chaque fois que possible.
Le traitement de la dystonie secondaire doit être guidée par l’objectif attendu : gain de fonction, qualité de vie… En règle générale, les traitements médicamenteux apportent un faible bénéfice aux enfants ayant des dystonies secondaires.2 Il faut savoir les limiter, ce d’autant qu’ils peuvent exposer à des effets indésirables notables (aggravation de l’hypotonie, impact négatif sur la cognition…). La L-dopa est recommandée en première intention si la cause de la dystonie n’est pas élucidée, de façon à ne pas passer à côté d’une dystonie dopa-sensible. On propose en deuxième intention les traitements anticholinergiques (trihexyphénidyle). Les injections de toxine botulique ont un intérêt dans les dystonies focales ou pour traiter un problème focal dans les dystonies généralisées. Ces enfants sont surtout aidés par la qualité de leur installation et de leur équipement (appareillage, tablette à pictogrammes, ordinateur avec logiciel de synthèse vocale…), par une éducation spécialisée et des soins réguliers de rééducation au sein d’un cadre adapté à leur handicap (ergothérapie, orthophonie, kinésithérapie…). La prise en charge des comorbidités (troubles du sommeil, de la déglutition, complications orthopédiques, douleur…) est primordiale, et contribue largement à préserver la qualité de vie et à limiter l’aggravation de la dystonie.
Dans une minorité de cas, la dystonie fait partie d’un syndrome dystonique génétiquement défini. Dans ce cadre, il est important de connaître les dystonies dopa-sensibles, disposant d’un traitement efficace. Elles se caractérisent par un début des mouvements anormaux dans l’enfance, avec une fluctuation diurne, une aggravation en fin de journée et une amélioration par le sommeil. Elles sont liées à un déficit enzymatique dans la voie de synthèse de la dopamine, entraînant une carence en neurotransmetteurs. Dans certaines formes, l’amélioration par la prise de L-dopa est spectaculaire. Le diagnostic repose sur le dosage des neurotransmetteurs dans le liquide céphalorachidien et l’analyse des gènes impliqués dans la synthèse de la dopamine.
Parmi les dystonies primitives non dopa-sensibles d’expression pédiatrique, les plus fréquentes impliquent les gènes torsine A (début dans l’enfance à un membre avec extension progressive vers une dystonie généralisée), le gène KMT2B (début dans l’enfance par les membres inférieurs et atteinte faciale affectant la parole), et le gène SGCE (survenue de secousses dans l’enfance affectant le plus souvent les membres supérieurs) [fig. 1 ]. La stimulation cérébrale profonde peut apporter un net bénéfice chez certains enfants atteints de formes à retentissement fonctionnel sévère.3
Le traitement de la dystonie secondaire doit être guidée par l’objectif attendu : gain de fonction, qualité de vie… En règle générale, les traitements médicamenteux apportent un faible bénéfice aux enfants ayant des dystonies secondaires.2 Il faut savoir les limiter, ce d’autant qu’ils peuvent exposer à des effets indésirables notables (aggravation de l’hypotonie, impact négatif sur la cognition…). La L-dopa est recommandée en première intention si la cause de la dystonie n’est pas élucidée, de façon à ne pas passer à côté d’une dystonie dopa-sensible. On propose en deuxième intention les traitements anticholinergiques (trihexyphénidyle). Les injections de toxine botulique ont un intérêt dans les dystonies focales ou pour traiter un problème focal dans les dystonies généralisées. Ces enfants sont surtout aidés par la qualité de leur installation et de leur équipement (appareillage, tablette à pictogrammes, ordinateur avec logiciel de synthèse vocale…), par une éducation spécialisée et des soins réguliers de rééducation au sein d’un cadre adapté à leur handicap (ergothérapie, orthophonie, kinésithérapie…). La prise en charge des comorbidités (troubles du sommeil, de la déglutition, complications orthopédiques, douleur…) est primordiale, et contribue largement à préserver la qualité de vie et à limiter l’aggravation de la dystonie.
Dans une minorité de cas, la dystonie fait partie d’un syndrome dystonique génétiquement défini. Dans ce cadre, il est important de connaître les dystonies dopa-sensibles, disposant d’un traitement efficace. Elles se caractérisent par un début des mouvements anormaux dans l’enfance, avec une fluctuation diurne, une aggravation en fin de journée et une amélioration par le sommeil. Elles sont liées à un déficit enzymatique dans la voie de synthèse de la dopamine, entraînant une carence en neurotransmetteurs. Dans certaines formes, l’amélioration par la prise de L-dopa est spectaculaire. Le diagnostic repose sur le dosage des neurotransmetteurs dans le liquide céphalorachidien et l’analyse des gènes impliqués dans la synthèse de la dopamine.
Parmi les dystonies primitives non dopa-sensibles d’expression pédiatrique, les plus fréquentes impliquent les gènes torsine A (début dans l’enfance à un membre avec extension progressive vers une dystonie généralisée), le gène KMT2B (début dans l’enfance par les membres inférieurs et atteinte faciale affectant la parole), et le gène SGCE (survenue de secousses dans l’enfance affectant le plus souvent les membres supérieurs) [
Tremblements
Ce sont des oscillations rythmiques involontaires, survenant le plus souvent chez l’enfant lors du maintien de la posture (exagération du tremblement physiologique, tremblement essentiel) ou à l’action (tremblement essentiel, tremblement cérébelleux).
Le tremblement essentiel chez l’enfant est très rare ; seuls 5 % de la population adulte ayant un tremblement essentiel ont un début dans l’enfance, ce tremblement touche alors plutôt les mains (là où les adultes peuvent avoir un tremblement du chef et/ou de la voix), impactant l’écriture, cet impact étant le motif de consultation en général.
Le tremblement de repos est rarissime chez l’enfant.
Parfois, l’enfant consulte pour un tremblement, et il s’agit en fait de secousses myocloniques (fig. 1 ) correspondant au déplacement d’un segment d’un point à un autre. On recherche en premier lieu une prise médicamenteuse pouvant être à l’origine du tremblement ou des myoclonies. Concernant les investigations, on propose un bilan biologique, avec dosage de la calcémie, bilan thyroïdien, cuprémie et céruléoplasmine (dépistage de la maladie de Wilson).4 L’imagerie cérébrale et l’enregistrement neurophysiologique du tremblement sont guidés par la clinique (naissance prématurée, présence de secousses...). Les diagnostics différentiels sont nombreux. L’évaluation du retentissement fonctionnel sur l’autonomie, la vie quotidienne, les activités scolaires et l’estime de soi (remarques récurrentes négatives…) est majeure. La prise en charge repose sur l’information claire du diagnostic et la mise en place d’aménagements et d’adaptations (tiers temps, ordinateur en classe pour la prise de notes), le recours à un traitement pharmacologique est rare chez l’enfant.
Le tremblement essentiel chez l’enfant est très rare ; seuls 5 % de la population adulte ayant un tremblement essentiel ont un début dans l’enfance, ce tremblement touche alors plutôt les mains (là où les adultes peuvent avoir un tremblement du chef et/ou de la voix), impactant l’écriture, cet impact étant le motif de consultation en général.
Le tremblement de repos est rarissime chez l’enfant.
Parfois, l’enfant consulte pour un tremblement, et il s’agit en fait de secousses myocloniques (
Mouvements anormaux paroxystiques
Mouvements transitoires du jeune enfant
Il s’agit de mouvements survenant dans la petite enfance, sans autres signes neurologiques associés et se résolvant spontanément dans la plupart des cas : accès de frissons (épisodes brefs de tremblements rapides du chef), myoclonies bénignes du nourrisson, favorisés par les émotions… Le visionnage sur vidéo du mouvement, l’anamnèse et l’examen clinique permettent de faire le diagnostic. Ce type de mouvement est important à reconnaître, de façon à éviter des investigations invasives inutiles ou des traitements inadaptés. Ils requièrent d’avoir soigneusement exclu les éventuels diagnostics différentiels (tumeur, épilepsie), et de suivre attentivement le développement de l’enfant.5
Tics
Ce sont des mouvements anormaux intempestifs brusques et rapides, survenant de façon involontaire et récurrente. Ils sont partiellement contrôlés par la volonté, précédés d’une sensation d’urgence à les faire, et succédés par un soulagement transitoire. Toutes ces caractéristiques sont importantes à relever en consultation, si l’enfant est en mesure de les exprimer. On peut aussi lui demander de montrer son tic en consultation. Les tics sont très fréquents chez les enfants. L’âge de survenue est classiquement au-delà de 3 ans. L’évolution est rémittente, avec des fluctuations. Le stress est un facteur favorisant, mais n’est pas la cause du tic. Une association de tics moteurs et vocaux durant plus d’un an sans période de rémission supérieure à 3 mois défini le syndrome de Gilles de la Tourette. Ce syndrome a tendance à s’améliorer de manière significative à l’âge adulte. Les comorbidités des tics avec d’autres troubles neurodéveloppementaux concernent plus de la moitié des enfants et sont donc importantes à dépister : trouble déficit de l’attention, trouble obsessionnel compulsif, trouble du spectre autistique.
La prise en charge repose essentiellement sur l’explication du trouble. Si l’enfant n’est pas gêné par ses tics, il est important de les tolérer, et de soutenir les parents dans ce sens. Si l’enfant est gêné par ses tics, la thérapie cognitivo-comportementale est efficace, et l’hypnose apparaît comme une approche très intéressante.6 Les traitements médicamenteux sont réservés aux formes sévères (aripiprazole notamment). Dans certains cas, la toxine botulique peut être une aide (douleurs, violents mouvements du cou pouvant léser la moelle épinière…).
La prise en charge repose essentiellement sur l’explication du trouble. Si l’enfant n’est pas gêné par ses tics, il est important de les tolérer, et de soutenir les parents dans ce sens. Si l’enfant est gêné par ses tics, la thérapie cognitivo-comportementale est efficace, et l’hypnose apparaît comme une approche très intéressante.6 Les traitements médicamenteux sont réservés aux formes sévères (aripiprazole notamment). Dans certains cas, la toxine botulique peut être une aide (douleurs, violents mouvements du cou pouvant léser la moelle épinière…).
Stéréotypies
Ce sont des mouvements involontaires répétitifs et rythmiques, sans objectif, volontiers favorisés par les émotions vives et l’excitation, et pouvant être interrompus par la distractibilité. Elles sont dites secondaires chez les enfants autistes, déficients intellectuels, s’intégrant parfois dans des syndromes génétiques (frottement de mains dans le syndrome de Rett…) et primaires chez les enfants sans handicap. Cette classification paraît obsolète, les stéréotypies pouvant être une co-occurrence de troubles subtils du neurodéveloppement (troubles praxiques, attentionnels…), ce qui incite à porter un regard attentif sur le neurodéveloppement de ces enfants.7 En outre, les enfants décrivent les sensations agréables que cela leur procure et apprennent, en grandissant, à privatiser leurs stéréotypies. Certains enfants ont des rythmies d’endormissement, parfois spectaculaires (l’enfant secoue bruyamment le lit). La prise en charge repose essentiellement sur la pose du diagnostic, évitant investigations et médicaments inadaptés. En cas d’impact sur le fonctionnement social, les approches comportementales sont intéressantes.
Mouvements oculaires anormaux
Certains d’entre eux nécessitent un avis neurologique, notamment les opsoclonies (secousses oculaires rapides multidirectionnelles [indication de rechercher un neuroblastome]), et l’apraxie oculomotrice (difficulté d’initiation des saccades oculaires, avec augmentation de leur latence, pouvant être acquise dans certaines pathologies neurodégénératives comme l’ataxie-télangiectasie, ou congénitale comme dans les malformations cérébelleuses).
Dyskinésies paroxystiques
Ce sont des accès intermittents de mouvements anormaux. On les classe selon leurs modalités de survenue. Les dyskinésies paroxystiques kinésigéniques sont déclenchées par le mouvement brusque, c’est le cas de celles liées aux mutations PRRT2 où les accès disparaissent avec de petites doses de carbamazépine. Les dyskinésies paroxystiques induites par l’exercice prolongé doivent faire évoquer en premier lieu un déficit en transporteur du glucose ou une dystonie dopa-sensible. L’hétérogénéité phénotypique du déficit en transporteur du glucose entraîne un sous-diagnostic alors que cette pathologie est importante à connaître car elle peut être traitée par des alternatives d’apports énergétiques autres que le glucose.8 Les dyskinésies paroxystiques non kinésigéniques ont des facteurs favorisants ou atténuants divers, indépendants du mouvement ou de l’effort. Ces facteurs varient en fonction de la cause.9
Mouvements anormaux liés à un trouble neurofonctionnel
Ces mouvements peuvent être très variés : sursauts, tremblements… C’est un diagnostic assez fréquent chez l’enfant, mais à poser avec prudence, une fois que l’on aura analysé attentivement l’histoire des troubles et pratiqué un examen clinique exhaustif, et après avoir pu observer le mouvement dans différents contextes et environnements (intérêt des vidéos). Un début brutal, une topographie inhabituelle, une variabilité importante des symptômes dans le temps, un retentissement fonctionnel sélectif ou peu proportionnel au mouvement anormal peuvent être des éléments cliniques en faveur de ce diagnostic. Ces mouvements sont entraînables (demander de battre la mesure au rythme de l’examinateur), suggestibles, et modifiables par la distraction (faire faire des tâches cognitives, comme le compte à rebours). Il ne s’agit en aucun cas de simulation, et il est important de poser ce diagnostic de façon positive, et non comme un diagnostic d’exclusion. La prise en charge peut être complexe, et l’explication bienveillante du diag- nostic reconnaissant la difficulté a une place cardinale dans le traitement. Il faut ensuite associer un suivi médical attentif à différentes approches, notamment corps-esprit (kinésithérapie, sophrologie, hypnose…), avec un suivi psychologique selon les cas (anxiété, présence d’un facteur déclenchant traumatique).
Références
1. Gras D, Jonard L, Roze E, et al. Benign hereditary chorea: phenotype, prognosis, therapeutic outcome and long term follow-up in a large series with new mutations in the TITF1/NKX2.1 gene. J Neurol Neurosurg Psychiatry 2012;83:956-62.
2. Roubertie A, Mariani LL, Fernandez-Alvarez E, Doummar D, Roze E. Treatment for dystonia in children. Eur J Neurol 2012;19:1292-9.
3. Mohammad SS, Paget SP, Dale RC. Current therapies and therapeutic decision making for childhood-onset movement disorders. Mov Disorder 2019;34:637-56.
4. Wilson R, Keener A. Movement disorders in children. Adv Pediatr 2018;65:229-40.
5. Bonnet C, Roubertie A, Doummar D, Bahi-Buisson N, Cochen de Cock V, Roze E. Developmental and benign movement disorders in childhood. Mov Disorder 2010;25:1317-34.
6. Flamand-Roze C, Célestin-Lhopiteau I, Roze E. Hypnosis and movement disorders: state of the art and perspectives. Rev Neurol 2016;172:530-6.
7. Gras D, Maes E. 100 idées pour mieux comprendre ce qu’est l’intelligence. Paris : Tom Pousse, 2018.
8. Gras D, Roze E, Caillet S, et al. Glut1 deficiency syndrome: an update. Revue Neurol (Paris) 2014;170:91-9.
9. Meneret A, Roze E. Paroxysmal movement disorders: an update. Rev Neurol (Paris) 2016;172:433-45.
2. Roubertie A, Mariani LL, Fernandez-Alvarez E, Doummar D, Roze E. Treatment for dystonia in children. Eur J Neurol 2012;19:1292-9.
3. Mohammad SS, Paget SP, Dale RC. Current therapies and therapeutic decision making for childhood-onset movement disorders. Mov Disorder 2019;34:637-56.
4. Wilson R, Keener A. Movement disorders in children. Adv Pediatr 2018;65:229-40.
5. Bonnet C, Roubertie A, Doummar D, Bahi-Buisson N, Cochen de Cock V, Roze E. Developmental and benign movement disorders in childhood. Mov Disorder 2010;25:1317-34.
6. Flamand-Roze C, Célestin-Lhopiteau I, Roze E. Hypnosis and movement disorders: state of the art and perspectives. Rev Neurol 2016;172:530-6.
7. Gras D, Maes E. 100 idées pour mieux comprendre ce qu’est l’intelligence. Paris : Tom Pousse, 2018.
8. Gras D, Roze E, Caillet S, et al. Glut1 deficiency syndrome: an update. Revue Neurol (Paris) 2014;170:91-9.
9. Meneret A, Roze E. Paroxysmal movement disorders: an update. Rev Neurol (Paris) 2016;172:433-45.
Dans cet article




Résumé
Les mouvements anormaux de l’enfant constituent un groupe très varié sur le plan phénoménologique, pouvant être reliés à des causes très distinctes, comprenant aussi bien des syndromes bénins transitoires que des maladies progressives. La clinique diffère sensiblement de celle de l’adulte, les troubles moteurs neurodéveloppementaux tels que tics et stéréotypies étant prédominants. La caractérisation précise du mouvement est le point clé de la stratégie diagnostique, en étant particulièrement attentif à l’anamnèse, aux symptômes associés, au développement psychomoteur, à l’évolution des symptômes, aux facteurs favorisants ou évitant la survenue du mouvement, et à l’interférence avec le mouvement volontaire. L’évaluation du retentissement fonctionnel est essentielle pour guider la prise en charge de l’enfant. Les investigations doivent s’attacher à identifier les causes potentiellement traitables de pathologie du mouvement. Chez l’enfant, l’abstention thérapeutique est la règle dans nombre de cas, pour limiter les effets indésirables et préserver au mieux les fonctions cognitives.