La mucoviscidose est la plus fréquente des affections héréditaires létales à transmission autosomique récessive dans les populations caucasiennes. Elle est liée à différentes mutations du gène codant la protéine CFTR (Cystic Fibrosis conductance Transmembrane Regulator) qui forme un canal chlorure transmembranaire. En 1989,1 la découverte de la première mutation a révolutionné le diagnostic et ouvert de nouvelles perspectives thérapeutiques.2 Le dépistage néonatal de la maladie est généralisé depuis 2002 (au 3e jour de vie, avec ceux de la phénylcétonurie, de l’hypothyroïdie, de l’hyperplasie des surrénales et de la drépanocytose) ; il a été associé à la création d’un réseau national de 47 centres de ressources et de compétences de la mucoviscidose (CRCM) qui prend en charge les patients et promeut la recherche.
La maladie touche près de 6 800 personnes en France, avec une incidence régionale variable (1/3 000 à 1/7 000). Les 75 000 patients mondiaux3 sont inégalement répartis en fonction de l’origine ethnique. Chez les Caucasiens, l’incidence est de 1/4 700 naissances, et seulement de 1/350 000 au Japon. La prévalence est estimée à 0,74/10 000 habitants dans l’Union européenne. Le sex-ratio est proche de 1, mais l’expression clinique est plus sévère chez les femmes. Alors que l’espérance de vie était de 7 ans en 1965, elle atteint les 50 ans pour les patients qui naissent aujourd’hui, grâce à l’amélioration des traitements et du suivi. La conséquence est que l’on voit apparaître des complications à moyen et long termes qui nécessitent une prise en charge spécifique, impérativement multidisciplinaire (cf. infra).
Physiopathologie
Le gène CFTR (en italique pour le distinguer de la protéine) est localisé sur le bras long du chromosome 7. Plus de 2 000 altérations sont connues (www.cftr2.org), dont le type et la fréquence varient selon l’origine ethnique. Si les 2 allèles portent chacun des mutations identiques : le patient est « homozygote » pour la mutation considérée. Si elles sont différentes, il est dit « hétérozygote composite ». Si un seul allèle est muté, il s’agit d’un « hétérozygote simple ».
Ces mutations sont groupées en 6 classes selon leurs conséquences sur la protéine CFTR :2
– classe I : altération de la production, d’où perte de fonction par défaut de synthèse ;
– classe II : défaut de maturation cellulaire et de transport vers la membrane plasmique. La protéine reste dans le cytoplasme où elle est dégradée. Ce sont les mutations les plus fréquentes, en particulier p.Phe508del ou F508del (70 % des patients) ;
– classe III : anomalie de régulation du canal chlorure : « gating mutations » pour les Anglo-Saxons. La protéine est correctement synthétisée et localisée à la membrane apicale mais ne peut être activée ou ne fonctionne pas ;
– classe IV : perturbation de la conduction ionique et des mécanismes d’ouverture et de fermeture du canal chlorure ;
– classe V : instabilité de l’ARN messager ;
– classe VI : altération de la stabilité de la protéine mature.
On distingue des mutations « sévères » (classe I, II et III) ou « modérées » (IV, V et VI). Cela a des implications thérapeutiques : de nouveaux médicaments ciblent, par exemple, celles de classe II et III (
Dépistage néonatal
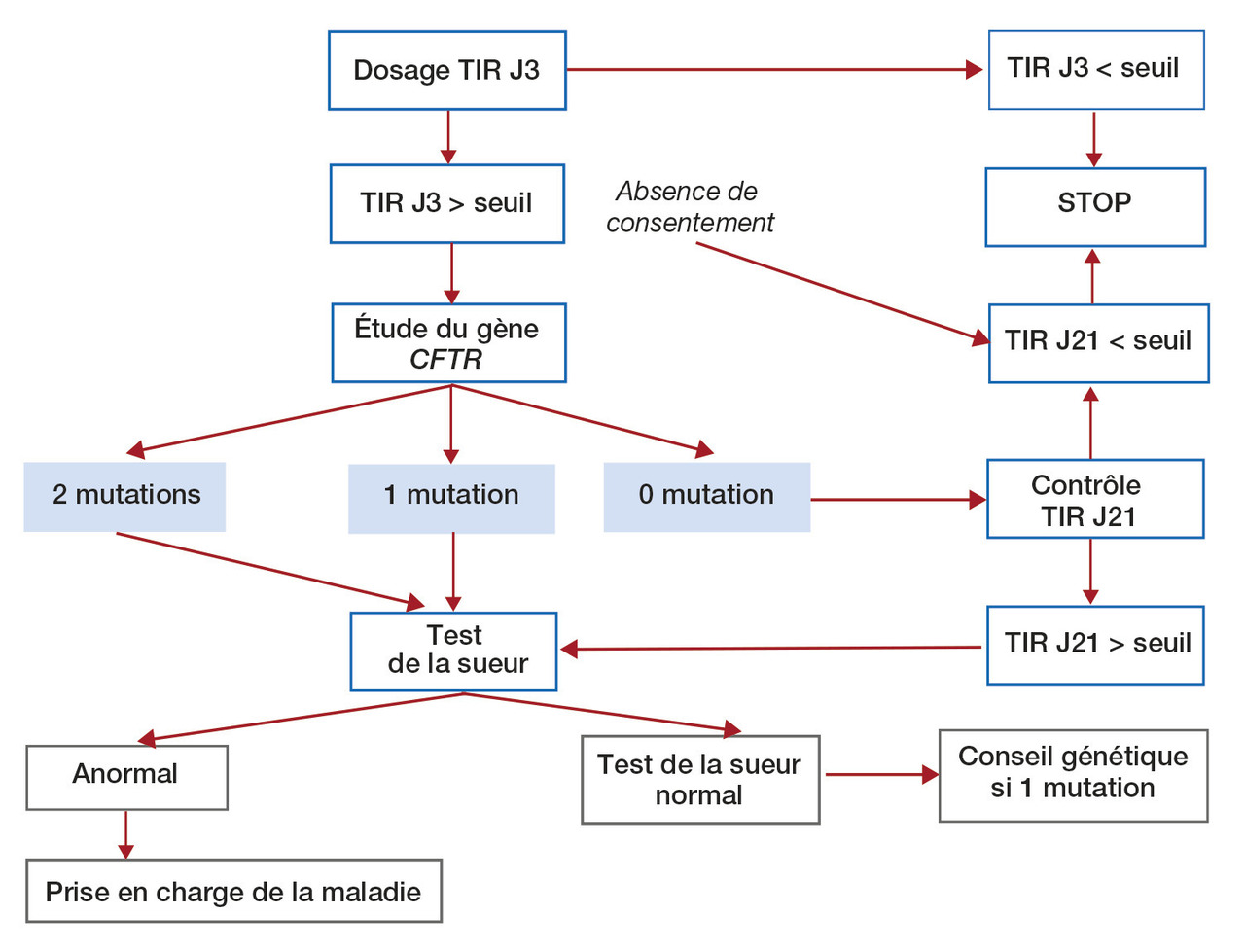
À la maternité, les parents sont informés de ses modalités et donnent ou pas leur consentement éclairé au génotypage de CFTR (
En pratique, les résultats sont transmis à l’Association française pour le dépistage et la prévention des handicaps de l’enfant (AFDPHE) qui contacte un CRCM si :
– 2 mutations identifiées (une dans chaque allèle) : le nouveau-né est alors convoqué par le centre le plus proche pour un test de la sueur qui confirme le diagnostic (
– 1 seule mutation identifiée (dans 1 seul allèle) : un test de la sueur est également pratiqué. S’il est anormal, il peut s’agir d’une mutation « hors kit de dépistage » et la mucoviscidose est confirmée. S’il est normal, l’enfant est porteur sain : un conseil génétique est recommandé.
Si aucune mutation identifiée ou absence de consentement parental : un contrôle de la TIR est réalisé à J21 par le médecin traitant ou la maternité. Lorsque la valeur dépasse le seuil de référence, l’enfant est adressé au CRCM pour un test de la sueur.
Résultats
Après les tests de la sueur dans les CRCM et, si nécessaire, analyse étendue du gène CFTR, 2 037 patients ont été identifiés : dont 1 743 formes « classiques » et 294 formes « frontières ». Ces dernières concernent les nouveau-nés ayant une TIR élevée à J3 et un test de la sueur intermédiaire, avec une mutation « modérée » ou aucune identifiée.7 À ce jour, la nécessité de les repérer fait débat. En effet, bien que la plupart restent peu ou pas symptomatiques, certaines peuvent évoluer vers des formes classiques.6
Il y a des faux négatifs. Leur nombre n’est qu’indicatif car le calcul nécessite du recul par rapport à l’année de naissance. Entre 2002 et 2014, en métropole, 138 diagnostics de mucoviscidose ont été faits malgré un dépistage négatif, certains dès la naissance (iléus méconial), d’autres plus tardivement devant des symptômes évocateurs ou à la suite du dépistage d’un membre de la fratrie.
Pronostic amélioré
Ses bénéfices nutritionnels à moyen et long terme sont confirmés par la majorité des études, en particulier celle mise en place prospectivement depuis 1985 au Wisconsin (États-Unis). Les enfants nés entre 1985 et 1994 ont été randomisés en « dépistage » ou en « contrôle ». Cent six diag-nostics ont été portés (56 dans le bras « dépistage » et 40 dans le groupe « témoin ») et les patients suivis jusqu’à l’âge de 18 ans. Une analyse très récente montre une meilleure croissance staturo-pondérale chez les dépistés, avec une taille finale significativement plus élevée.8
Les bénéfices respiratoires sont plus difficiles à démontrer en raison du manque de sensibilité des outils de mesure. À l’âge de 2 ans, dans le groupe témoin, 2 fois plus d’enfants ont des atteintes radiologiques irréversibles.9 Cependant, après 10 ans de suivi, cette différence s’inverse. Ce qui pourrait s’expliquer par une colonisation plus précoce des voies aériennes par Pseudomonas aeruginosa : conséquence potentielle de l’insuffisance de ségrégation des patients selon leur statut bactériologique (les malades les plus infectés doivent être examinés ou pris en charge en dernier).
Bien que le recul soit trop court pour conclure formellement, les données sont en faveur du dépistage, avec moins de décès précoces chez les patients dépistés.
Expression clinique
La mucoviscidose prédomine au poumon, au tube digestif et ses annexes (pancréas, voies biliaires). Sont également concernés les glandes sudoripares, l’appareil génital et le système ostéo- articulaire. Les phénotypes cliniques varient selon la mutation en cause.
L’atteinte pulmonaire est le facteur principal de morbi-mortalité. La déshydratation des sécrétions bronchiques diminue la clairance mucociliaire favorisant l’obstruction chronique des bronchioles par un mucus épais. Cela entraîne une inflammation bronchique excessive et des infections à répétition, particulièrement à germes opportunistes : Staphylococcus aureus et Haemophilus influenzae, puis Pseudomonas aeruginosa. Cette bactérie, naturellement résistante à de nombreux antibiotiques, devient peu à peu l’agent pathogène majeur des voies respiratoires.10 Les symptômes initiaux associent des bronchites récidivantes à une toux grasse, productive et perannuelle. L’évolution se fait par poussées, ou exacerbations, avec dégradation progressive de l’épithélium qui aboutit à une insuffisance respiratoire (cf. infra et
L’insuffisance pancréatique exocrine (IPE, défaut de production des enzymes pancréatiques) survient chez 85 % des patients par accumulation de bouchons muqueux dans les canaux excréteurs qui provoquent la dégénérescence du tissu pancréatique. Il faut la rechercher systématiquement en dosant l’élastase fécale : inférieure à 200 μg/g de selles, elle est pathologique. Si le taux est normal, on répète le dosage, surtout s’il y a stéatorrhée et/ou trouble de la croissance.
La conséquence est une maldigestion lipidoprotidique et une malabsorption (avec carence en vitamines ADEK et acides gras essentiels).12 La fonction exocrine (déficiente ou conservée) dépend du type de mutations de CFTR. Les patients insuffisants pancréatiques sont ceux ayant 2 altérations dites « sévères » (classe I, II ou III) et un phénotype grave de la maladie. L’IPE n’a de retentissement clinique que lorsque 90 % du pancréas est détruit.
L’insuffisance pancréatique endocrine, ou diabète de la mucoviscidose, apparaît après l’âge de 10 ans et touche près d’un patient sur trois après 30 ans.12 Il s’agit d’un diabète à la fois insulinoprive (diabète de type 1) parce que secondaire à la destruction des îlots de Langerhans et insulinorésistant (de type 2), la résistance tissulaire faisant suite aux infections récurrentes et aux traitements par corticoïdes. Ses facteurs de risque sont l’âge, le sexe féminin, les mutations «sévères» de CFTR, l’intensité de l’IPE, la cirrhose hépatique et l’insuffisance respiratoire sévère. Il faut le rechercher systématiquement en réalisant annuellement à partir de l’âge de 10 ans une hyperglycémie provoquée par voie orale (HGPO) et un dosage de l’hémoglobine glyquée (HbA1c).
Les troubles digestifs peuvent être immédiats, dès la naissance, se manifestant par un iléus méconial (environ 15 % des patients) : occlusion intestinale basse avec absence d’émission du méconium dans les premiers jours de vie. Plus tard, des syndromes d’obstruction intestinale distale (SOID) équivalents à des iléus méconiaux surviennent par obstruction de l’intestin grêle.13 Le reflux gastro-œsophagien est fréquent.
L’atteinte hépatique se résume dans 30 % des cas à une hépatomégalie. Elle évolue vers une cirrhose13 (2 à 5 %) par obstruction des voies biliaires intra- et extrahépatiques, compliquée ou pas d’hypertension portale.
Les manifestations génitales se traduisent chez la femme par une hypofertilité due aux modifications de la glaire cervicale. Chez l’homme, l’agénésie bilatérale des canaux déférents entraîne une azoospermie et une stérilité.
Les glandes sudoripares produisent un excès de chlorure de sodium dans la sueur. Cette perte de sel peut entraîner une déshydratation aiguë lors d’une d’exposition prolongée à la chaleur ou lors d’efforts physiques importants.
La déminéralisation osseuse est fréquente, en particulier chez les adultes. L’ostéopénie touche près de 85 % des patients et l’ostéoporose 13 à 57 %.14 Nombreux sont les facteurs favorisants : dénutrition, inflammation, déficits en vitamines, corticothérapie et sédentarité.
Traitement de fond
La prise en charge musculo-squelettique comporte la prévention et la rééducation des troubles engendrés par l’atteinte respiratoire, en particulier la cyphose dorsale et les contractures musculaires. La rééducation périnéale pour les problèmes urinaires fréquents chez les femmes s’inscrit dans ce cadre.
Gestion nutritionnelle
Une dénutrition est fréquente par diminution des ingestats (IPE, encombrement bronchique, douleurs abdominales et prises médicamenteuses), et augmentation des pertes secondaire à l’inflammation et à l’accroissement du travail musculaire respiratoire. L’enrichissement est le plus souvent nécessaire avec l’aggravation de la maladie : entre 120 et 150 % des apports nutritionnels conseillés chez les enfants de 2 ans et plus (
L’insuffisance pancréatique exocrine nécessite une supplémentation en extraits pancréatiques oraux à chaque repas, en fonction des symptômes digestifs et de la croissance. La malabsorption des vitamines liposolubles (A, D, E, K et bêtacarotène) et parfois de vitamine B12 et de zinc, sélénium et magnésium doit être compensée (compléments vitaminiques et oligoéléments adaptés au cas par cas).
Atteinte bronchopulmonaire
L’administration de mucolytiques fluidifie les sécrétions. Les aérosols de désoxyribonucléase recombinée humaine (rhDNase ou dornase alfa ou Pulmozyme) hydrolysent de grandes quantités d’ADN issu notamment des polynucléaires et des bactéries. Cela diminue la viscosité du mucus, améliore la fonction pulmonaire et réduirait même l’inflammation. L’effet osmotique de l’inhalation de sérum salé hypertonique favorise l’hydratation du mucus et augmente la clairance mucociliaire.
L’antibiothérapie doit être précoce, débutée dès la première exacerbation pulmonaire, accompagnée d’ECBC par ailleurs réalisés en routine. Ils identifient les germes en cause et évaluent leur sensibilité en vue d’une prescription adaptée. Dans un premier temps, celle-ci vise principalement S. aureus et H. influenzae, souvent par voie orale. La primocolonisation à P. aeruginosa est traitée, selon la clinique, per os ou IV en association à un antibiotique inhalé. Les exacerbations sur infection chronique sont gérées par voie veineuse. Pour les autres infections, le choix thérapeutique est fonction du germe, des symptômes et de l’antibiogramme.
Très rapidement s’installe un cercle vicieux impliquant inflammation, infections et libération de dérivés oxydants. C’est pourquoi les anti- inflammatoires et anti-oxydants sont nécessaires. Les AINS sont peu precrits car ils nécessitent des doses importantes, générant trop d’effets secondaires gastriques et rénaux. Les corticoïdes par voie systémique pourraient lutter contre l’inflammation pulmonaire mais, étant potentiellement délétères, ils ne sont prescrits que dans des cas précis, tels que les aspergilloses bronchopulmonaires allergiques (ABPA). Les corticoïdes inhalés n’ont pas montré d’efficacité. Actuellement, le traitement anti-inflammatoire de choix est l’azithromycine au long cours à faible dose.15 Sa tolérance clinique doit être surveillée, associée aux ECBC qui traquent notamment l’émergence d’une résistance.
Les vaccins obligatoires et recommandés sont tous à faire. La vaccination antigrippale est recommandée dès l’âge de 6 mois ; avant cet âge, il est préconisé de vacciner l’entourage proche.
Modalités du suivi
– pédiatre, pneumologue, gastro-entérologue ou interniste ;
– kinésithérapeute, infirmier(e) coordonnateur (trice), diététicien(ne), psychologue, assistante sociale, secrétaire, généticien clinicien ;
– autres professionnels hospitaliers impliqués tels que le microbiologiste référent et le biologiste responsable du test de la sueur, le généticien moléculaire ;
– interlocuteurs libéraux (médecin traitant, kinésithérapeute, pharmacien, infirmière) en charge du patient.
Le Programme national de diagnostic et de soins (PNDS, HAS 2017)16 a déterminé la fréquence et les modalités du suivi : au moins tous les 2 mois jusqu’à l’âge de 1 an, puis tous les 2 à 3 mois ensuite avec au moins une consultation trimestrielle et un bilan annuel complet (hospitalisation de jour ou de courte durée) pour tous les patients.
Dans l’intervalle, le médecin traitant gère les pathologies intercurrentes, en relation avec un médecin du CRCM si nécessaire.
À chaque visite de suivi en centre référent, l’interrogatoire et l’examen clinique évaluent particulièrement l’état respiratoire et digestif. Le kinésithérapeute éduque le patient, dégage les voies respiratoires et fait un ECBC. La spirométrie est systématique dès que le patient est en âge de la réaliser correctement, généralement à partir de 5-6 ans. L’infirmier(e) coordinateur(rice) organise les soins au centre et à domicile en partenariat avec l’infirmier(e) libéral(e), le pharmacien et le prestataire de matériel.
Au moins une fois par an : le(la) diététicien(ne) fait un bilan nutritionnel complet et vérifie l’éducation alimentaire. Une consultation avec le psychologue et l’assistante sociale est proposée, ainsi que l’avis des spécialistes d’organes selon l’âge et les complications.
Les examens complémentaires dépendent des atteintes et de leur sévérité (annexe 5 du PNDS).16 Pour tous les patients et en dehors des compli- cations, sont réalisés au moins 1 fois par an : radiographie du thorax, échographie abdominale et bilan sanguin (marqueurs de l’inflammation, glycémie, bilan phosphocalcique, rénal et entéro- hépatique).
L’éducation thérapeutique du patient et des proches est structurée en 4 étapes : diagnostic éducatif, programme personnalisé, mise en œuvre de séances et évaluation. Elle développe entre autres : physiopathologie de la maladie, thérapeutiques et effets indésirables, signes d’aggravation motivant une consultation en urgence, diététique, drainage bronchique, désencombrement… Ces rendez-vous impliquant médecins, infirmières, diététiciennes, kinési-thérapeutes, psychologues se déroulent en individuel ou en groupe, généralement au CRCM du patient.
Complications respiratoires
Les exacerbations pulmonaires sont le plus souvent dues à des surinfections bronchiques, à des hémoptysies ou des pneumothorax, à des décompensations diabétiques. Il n’y a actuellement pas de consensus pour les définir. On retient l’association de critères cliniques (asthénie, perte d’appétit, amaigrissement, aggravation de la toux, modification des expectorations, de la dyspnée, de la tolérance à l’effort, de l’auscultation pulmonaire, hémoptysie) et paracliniques (diminution de la saturation en oxygène ou du VEMS, majoration des anomalies de l’imagerie thoracique). Leur dépistage précoce se fait grâce à la collaboration entre le CRCM et le médecin traitant et/ou le kinésithérapeute de ville, et par l’éducation du patient et de ses proches.
Du fait de leurs conséquences néfastes (1 patient sur 4 ne récupère pas sa fonction respiratoire antérieure), la prise en charge est énergique : antibiothérapie probabiliste (adaptée secondairement aux résultats de l’ECBC) et optimisation de la kinésithérapie respiratoire. La gestion ambulatoire ou l’hospitalisation se discutent en fonction de la sévérité de l’épisode.
Les hémoptysies sont plus souvent des crachats hémoptoïques que des rejets massifs. Leur incidence annuelle est estimée à 5 %.3 Plus fréquentes chez les adultes avec une maladie avancée, elles majorent la mortalité et justifient une consultation en urgence au CRCM. La prise en charge, variable, comprend une hospitalisation de surveillance et une antibiothérapie probabiliste. Une artériographie bronchique avec embolisation peut se discuter.
Les pneumothorax sont une complication rare (incidence annuelle estimée à 0,9 %)3 qui survient plutôt chez les patients au stade avancé de la maladie. Ils aggravent aussi la mortalité. La consultation en urgence au CRCM est nécessaire. L’hospitalisation permet une antibiothérapie probabiliste associée ou pas à un drainage simple, une symphyse médicale ou chirurgicale (vidéo- assistée ou par thoracotomie).
L’insuffisance respiratoire chronique est affirmée sur un enregistrement de la saturation pulsée en oxygène (SpO2) réalisé préférentiellement la nuit pendant 8 heures, et associé à celui des pressions partielles artérielles en O2 (PaO2) et en CO2 (PaCO2). L’oxygénothérapie au long cours à domicile (OLD) est indiquée :
– si à l’éveil, la PaO2 est inférieure à 55 mmHg ou comprise entre 56 et 59 mmHg, avec une hypertension artérielle pulmonaire (HTAP) ;
– si pendant le sommeil, la SpO2 est inférieure à 90 % sur plus de 10 % de l’enregistrement. L’oxygénothérapie temporaire et/ou de déambulation peut être nécessaire, par exemple au cours d’une exacerbation. La ventilation non invasive (VNI) est indiquée en cas d’hypercapnie et/ou de dégradation respiratoire. La transplantation pulmonaire est discutée en cas d’insuffisance respiratoire sévère (
Adobe stock
1. Nouvelles thérapeutiques
L’ivacaftor (Kalydeco) est indiqué chez les sujets de plus de 2 ans avec au moins une mutation de classe III suivante : G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P et G1349D. Ce qui correspond à 2-3 % des patients. Posologie : un comprimé de 150 mg 2 fois par jour, à prendre avec un repas riche en graisses. L’autorisation de mise sur le marché (AMM) date de 2012 pour la mutation G551D et de 2015 pour les 8 autres mutations plus rares. Cette molécule permet l’ouverture du canal chlorure, améliore le VEMS de presque 10 % en moyenne, raréfie les exacerbations respiratoires et favorise la prise de poids. Le bilan hépatique est recommandé 1 mois après l’initiation du traitement, tous les 3 mois la première année de prescription puis 1 fois par an. L’examen ophtalmologique avant sa mise en place élimine une cataracte.
L’association de l’ivacaftor et du lumacaftor (facilitant l’acheminement de CFTR à la membrane cellulaire) améliore modérément le VEMS et diminue les exacerbations respiratoires chez les plus de 12 ans homozygotes pour la mutation F508del, soit environ 1 500 malades potentiels en France. Commercialisé sous le nom d’Orkambi (200 mg de lumacaftor + 125 mg d’ivacaftor) depuis fin 2015, ce traitement est prescrit à la dose de 2 comprimés matin et soir. Une nouvelle molécule, le tezacaftor, pourrait prochainement remplacer le lumacaftor dans cette association, car un essai publié récemment a montré une efficacité supérieure.4
2. Colonisation et infection bronchiques : inéluctables
L’infection bronchique bactérienne survient en général dès les premières semaines de vie. Initialement aiguë, elle devient chronique malgré l’antibiothérapie (qui permet l’amélioration clinique et la réduction transitoire de la charge bactérienne). L’examen cytobactériologique des crachats (ECBC) est systématique au moins 4 fois par an. À l’adolescence, on recherche des mycobactéries atypiques au moins 2 fois par an.
On distingue 3 stades infectieux dont les thérapeutiques diffèrent.
• La primo-infection ou primo-colonisation désigne la découverte d’une première bactérie sans anticorps réactionnel (surtout s’il s’agit de P. aeruginosa).
• L’infection intermittente correspond à une éradication transitoire, de durée variable mais supérieure à 6 mois (ou < 50 % des prélèvements de l’année) sous réserve d’ECBC à la fréquence recommandée.
• L’infection ou colonisation chronique est définie par la présence du germe pendant au moins 6 mois, sur 3 prélèvements à 1 mois d’intervalle (ou > 50 % des prélèvements de l’année), et/ou par un taux significatif d’anticorps (essentiellement pour P. aeruginosa).
3. Critères de discussion d’une transplantation pulmonaire
• VEMS inférieur à 30 % malgré une thérapie médicale optimale
• Déclin rapide de la fonction respiratoire surtout s’il est associé à des facteurs de mauvais pronostic comme la dénutrition ou le diabète
• Exacerbations fréquentes avec mauvaise réponse aux antibiothérapies intraveineuses
• Pneumothorax réfractaires ou hémoptysies massives et récurrentes
• Hospitalisation en soins intensifs pour exacerbation
• Dépendance à l’oxygène et/ou à la ventilation non invasive
Vaincre la mucoviscidose, association pionnière
Dédiée au soutien et à l’information des malades et de leur famille, l’association participe activement au Registre français de la mucoviscidose labellisé en 2006. En 2016, l’intégration des données de l’AFDPHE, de Muco-CFTR et de CépiDc (Inserm) a amélioré qualité et exhaustivité du recueil, aboutissant à un recensement de 6 757 patients dont 55 % d’adultes (11,6 % de plus de 40 ans contre 3,6 % en 2006).
Vaincre la mucoviscidose offre aussi un appui incontournable à la recherche. Favorisant la collaboration entre partenaires nationaux et internationaux, elle finance sur appel à projets, organise des colloques et anime des réseaux spécifiques. Le financement direct est le plus gros budget de cette mission. Une sélection rigoureuse des projets optimise l’usage des dons (5,8 millions d’euros en 2016) : qualité scientifique et objectif thérapeutique, équipes impliquées, pertinence par rapport à la maladie, adéquation entre devis et travaux envisagés, validation scientifique, choix stratégiques.
En 2017, Vaincre la mucoviscidose (www.vaincrelamuco.org) a financé 68 projets et soutient de grandes actions, telle COLT (avec l’association Grégory Lemarchal) qui tente de mieux contrôler le Chronic Lung Allograft Dysfunction after lung transplantation (CLAD). Cette destruction irréversible du transplant pulmonaire par rejet immun survient chez 35 à 50 % des greffés dans les 2 à 5 ans.
2. Corvol H, Taytard J, Tabary O, et al. Challenges of personalized medicine for cystic fibrosis. Arch Pediatr 2015;22:778-86.
3. Bellis G, Delhillotte C, Lemonnier L. Registre français de la mucoviscidose. Bilan des données 2015.
4. Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017;377:2013-23.
5. HAS. Mucoviscidose: Protocole national de diagnostic et de soins pour une maladie rare. Guide - Affection de longue durée. Novembre 2006.
6. AFDPHE. Bilan d’activités 2014.
7. Munck A, Mayell SJ, Winters V, et al.; ECFS Neonatal Screening Working Group. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J Cyst Fibros 2015;14:706-13.
8. Zhang Z, Lindstrom MJ, Farrell PM, Lai HJ.; Wisconsin Cystic Fibrosis Neotnatal Screening Group. Pubertal Height Growth and Adult Height in Cystic Fibrosis After Newborn Screening. Pediatrics 2016;137:e20152907.
9. Farrell PM, Kosorok MR, Rock MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics 2001;107:1-13.
10. Marguet C, Lemee L, Morisse-Pradier H, Couderc L.Infection cystic fibrosis: Up-to-date. Arch Pediatr 2016;23: 12S33-12S38.
11. Sermet-Gaudelus I, Munck A, Rota M, et al. French guidelines for sweat test practice and interpretation for cystic fibrosis neonatal screening. Arch Pediatr 2010;17: 1349-58.
12. Kessler L, Abély M. Pancreatic infringement exocrine and endocrine in cystic fibrosis. Arch Pediatr 2016;23: 12S21-12S32.
13. Debray D, Mas E, Munck A, Gerardin M, Clouzeau H. Liver disease, gastrointestinal complications, nutritional management and feeding disorders in pediatric cystic fibrosis. Arch Pediatr 2016;23:12S15-12S20.
14. Sermet-Gaudelus I, Nove-Josserand R, Loeille GA, et al. Recommendations for the management of bone demineralization in cystic fibrosis. Arch Pediatr 2008;15: 301-12.
15. HAS. Mucoviscidose: Protocole national de diagnostic et de soins pour une maladie rare. Guide - Affection de longue durée. Septembre 2017.
16. Corvol H, Taytard J, Thouvenin G, et al. Why use long-term macrolide therapy in pediatric pulmonology? Arch Pediatr 2014;21:314-21.
 Encadrés
Encadrés