Aujourd’hui, il n’est pas rare en réunion de concertation pluridisciplinaire de discuter de cas évoluant depuis plus de 10 ans, voire 15 ans. La maladie reste cependant incurable, et la rechute, à quelques exceptions près, inévitable.
Autre ombre au tableau, certaines formes à « haut risque » cytogénétique (10 à 15 % des cas) gardent malgré les progrès thérapeutiques des survies particulièrement courtes, de l’ordre de 3 ans, parfois moins.
Physiopathologie
Le MM est une maladie du plasmocyte, cellule terminale de la différenciation lymphocytaire B, capable de sécréter des anticorps (ou immuno-globulines). En situation normale, ces derniers diffèrent tous d’un plasmocyte à l’autre, grâce à des phénomènes combinatoires impliquant divers segments de gènes, à l’origine d’un répertoire immun quasi infini. Ils assurent ainsi la reconnaissance d’un très grand nombre d’antigènes, en particulier microbiens.
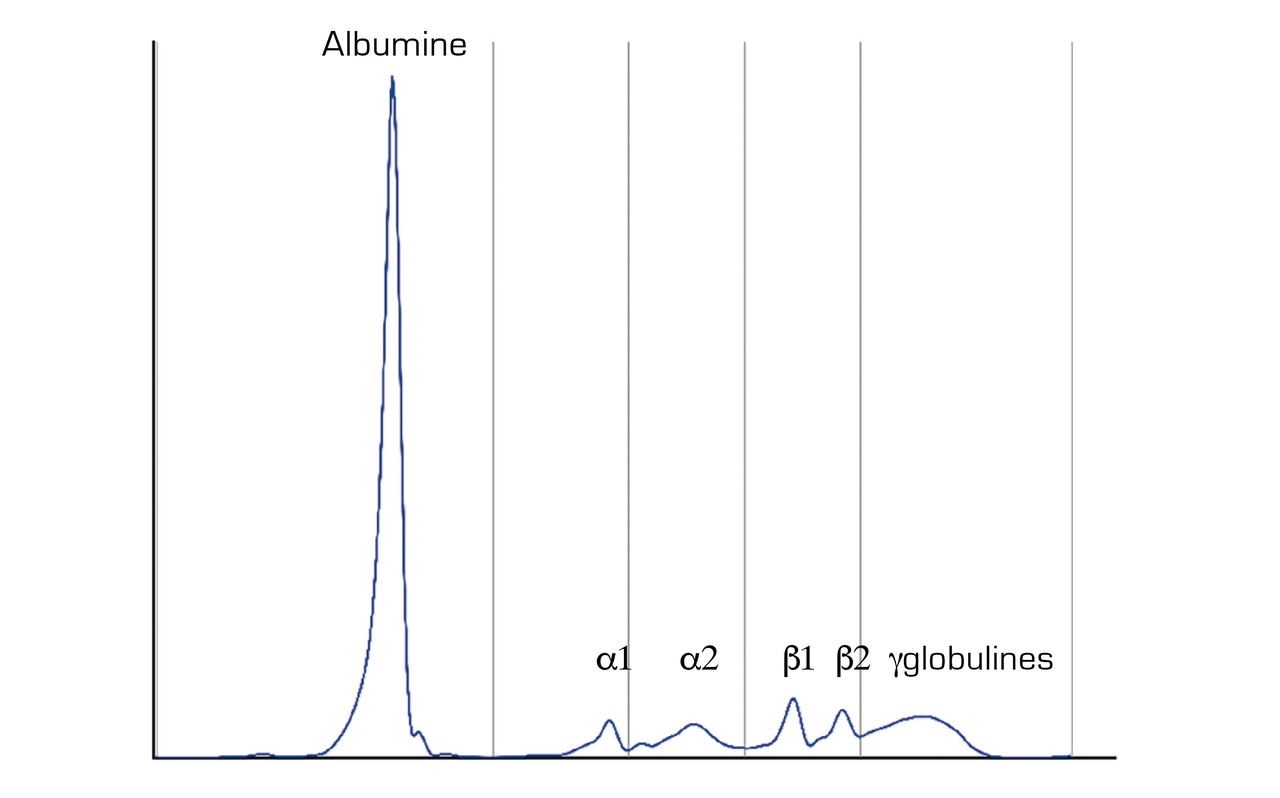
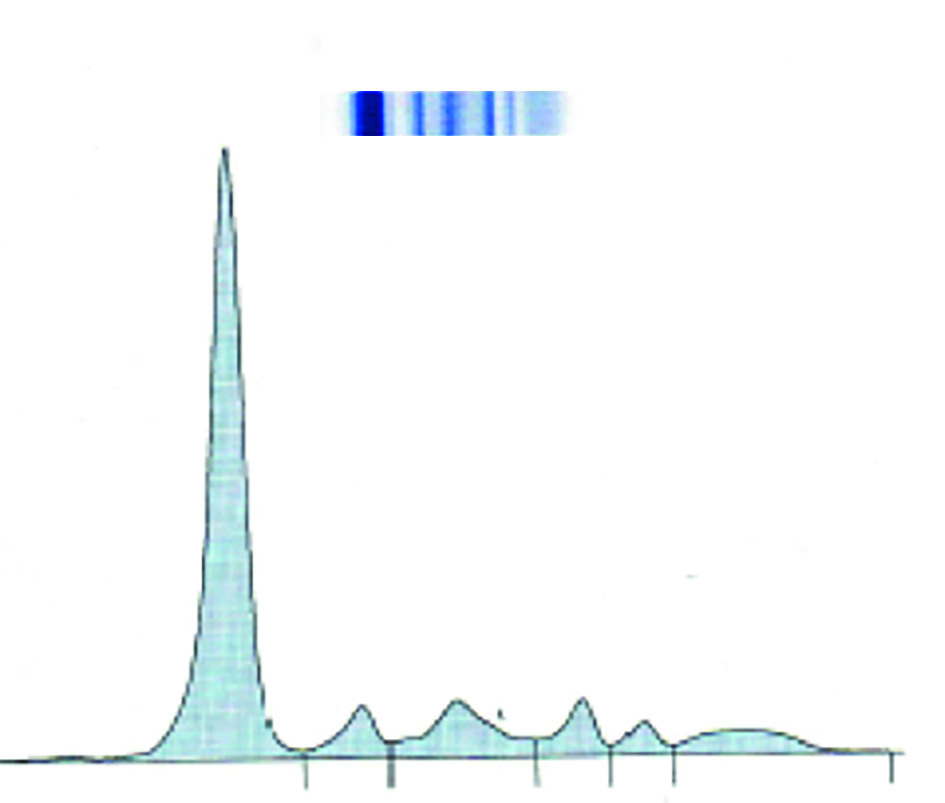
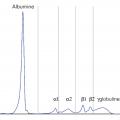
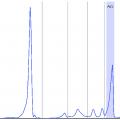
À l’électrophorèse des protéines sériques (EPS), ils migrent tous de manière différente, donnant, dans la zone des gammaglobulines, une forme étalée en « colline » illustrant cette diversité (
Conséquences de la prolifération plasmocytaire
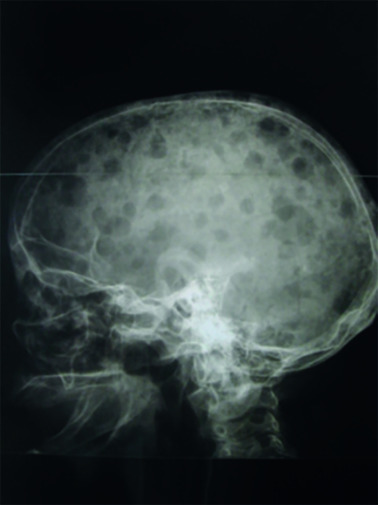
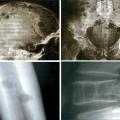
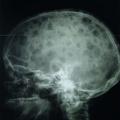
Les plasmocytes myélomateux désorganisent l’homéostasie du stroma médullaire, en activant les ostéoclastes et en inhibant les ostéoblastes. Ce déséquilibre entre résorption et fabrication osseuse conduit à l’apparition de lésions ostéolytiques. Elles sont classiquement « à l’emporte-pièce », principalement dans les os riches en moelle : crâne (
Une hypercalcémie par ostéolyse maligne est alors possible. Elle s’exprime par des symptômes digestifs (nausées, douleurs abdominales), un syndrome polyuro-polydipsique, des troubles neurologiques (confusion, troubles du langage).
Probablement par un assemblage défectueux des différentes parties de l’immunoglobuline dans les plasmocytes tumoraux, certains MM, dits « à chaînes légères », ne produisent que de grandes quantités de fragments d’Ig monoclonale (absence d’anticorps entier). Ces chaînes légères peuvent précipiter dans les tubules rénaux et entraîner une insuffisance rénale, parfois aiguë : c’est la tubulopathie myélomateuse (ou néphropathie à cylindres myélomateux). Elle est souvent déclenchée ou aggravée par la déshydratation, l’hypercalcémie, les infections et certains médicaments (AINS et produits de contraste iodés en particulier).1
Tous les phénomènes qui précèdent sont résumés par l’acronyme « CRAB » (
En outre, la répression concomitante de la synthèse des Ig polyclonales (par un mécanisme mal connu) est à l’origine d’un déficit immun humoral profond, rendant les malades particulièrement sensibles aux germes encapsulés (plus d’opsonisation possible par les anticorps permettant une destruction cellulaire efficace) : le pneumocoque notamment, Haemophilus influenzae et des bacilles à Gram négatif.
Découverte et diagnostic
Son incidence en Europe est de 4,5 à 6 cas pour 100 000. En France, elle était de 4 888 cas en 2015 (52,4 % d’hommes).
Les douleurs osseuses sont le mode de découverte le plus fréquent : rachidiennes en particulier, d’horaire inflammatoire (penser à un myélome avant de prescrire des AINS), fractures costales ou parfois fracture pathologique.
Mentionnons la révélation, classique, à l’occasion d’une complication infectieuse, en particulier une pneumonie franche lobaire aiguë, parfois une méningite à pneumocoque.
Quelles explorations ?
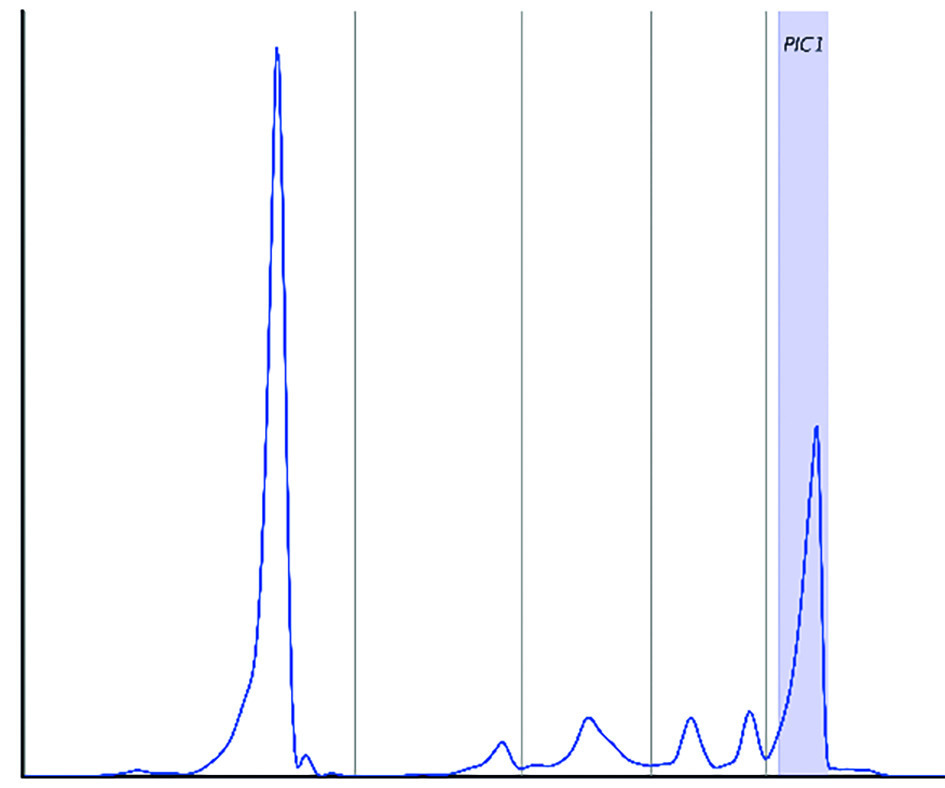
Cependant, dans 10 à 20 % des cas environ, seule la chaîne légère de l’Ig monoclonale est produite (MM à chaînes légères). On ne retrouve alors pas de pic à l’EPS mais une hypogammaglobulinémie (
Point important, cette protéinurie n’est pas souvent détectée à la bandelette. Il faut donc bien demander une recherche de protéinurie (au mieux sur les urines de 24 heures) et surtout, élément fondamental, une électrophorèse des protéines urinaires, afin de confirmer qu’il s’agit bien d’une protéinurie de surcharge tubulaire.
Si la protéinurie est glomérulaire (c’est-à-dire composée essentiellement d’albumine), on doit alors évoquer une néphropathie glomérulaire en lien (amylose, maladie des dépôts notamment) ou pas (néphropathie diabétique, néphro-angiosclérose) avec la gammapathie monoclonale.2
Pour compléter les explorations, on demande des radiographies simples des os (pas besoin de scintigraphie osseuse, les lésions du myélome étant peu ou pas fixantes).
Explorations en milieu spécialisé
– immunofixation sanguine (et urinaire pour les formes à chaînes légères), qui confirme le caractère monoclonal et détermine l’isotype de l’Ig. Il s’agit le plus souvent d’une IgG pour la chaîne lourde, moins fréquemment d’une IgA (10 à 15 %), très rarement d’une IgD. La chaîne légère est kappa dans deux tiers des cas, lambda pour le dernier tiers ;
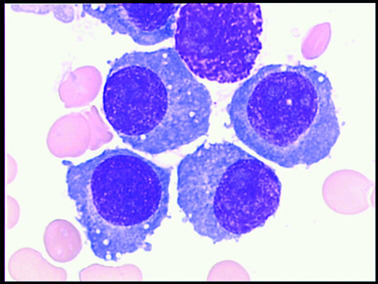
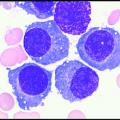
– myélogramme montrant la prolifération plasmocytaire, avec des cellules souvent dystrophiques.
Dans les formes à chaînes légères, on s’aide du dosage par néphélémétrie des chaînes légères libres sériques (à ce jour non remboursé en laboratoire de ville). Il n’est pas utile de les doser dans les urines (valeur non interprétable).
En cas de suspicion d’atteinte neurologique (déficit sensitivomoteur aux membres, douleurs en ceinture évoquant un syndrome lésionnel), l’IRM rachidienne recherche une épidurite et/ou des tassements vertébraux caractéristiques en galette, responsables d’une compression médullaire ou des racines. Le TEP-scan est de plus en plus utilisé, mais davantage pour évaluer la réponse au traitement qu’à visée diagnostique.
Évaluer le pronostic
Diagnostic différentiel
En revanche, il ne faut pas méconnaître la possibilité d’une gammapathie dite de signification clinique (MGCS), où l’Ig monoclonale en elle-même, même à très faible taux peut, par ses propriétés physicochimiques, entraîner des atteintes d’organes parfois sévères : amylose AL (à évoquer devant une macroglossie, des signes d’insuffisance cardiaque, une protéinurie glomérulaire), cryoglobulinémie (purpura vasculaire, douleurs articulaires).4
Enfin, une Ig monoclonale peut être satellite de certains lymphomes, d’une leucémie lymphoïde chronique. On doit donc également examiner les aires ganglionnaires et rechercher une splénomégalie.
Le risque de progression d’une MGUS vers un MM est de 1% par an, très dépendant bien-sûr du taux de l’Ig monoclonale. La surveillance est essentiellement fonction de celui-ci, et également du terrain sous-jacent (âge, comorbidités). Dans tous les cas, un contact entre le médecin traitant et un hématologue est conseillé pour définir au mieux les modalités de l’expertise et du suivi.
La découverte d’une Ig monoclonale de taux plus important (> 20-30 g/L) associée à une infiltration plasmocytaire significative au myélogramme (10 % ou plus), mais sans critère CRAB, définit le myélome multiple indolent (smolderingmyeloma), qui peut rester quiescent pendant des années, comme il peut évoluer vers un MM symptomatique.5 La surveillance se fait ici tous les 3 à 6 mois, dans tous les cas en milieu spécialisé. L’IRM rachi-dienne est particulièrement utile comme outil prédictif d’évolution vers un myélome symptomatique. On y recherche des anomalies de signal au niveau de la moelle osseuse, correspondant à des lésions infraradiologiques, non détectées sur les radiographies simples.
à ce jour, en l’absence de traitement curateur et étant donné les différences de profils évolutifs, les MM indolents ne relèvent pas, dans la pratique courante, d’un traitement antitumoral. Des recherches sont actuellement menées pour discriminer au mieux les formes amenées à évoluer de celles qui resteront indolentes.
Principes du traitement
Pour les sujets jeunes
Chez les patients répondeurs (80-90 %), on procède ensuite à un recueil de cellules souches médullaires prélevées dans le sang périphérique par cytaphérèse, en général à la suite d’une chimiothérapie qui les mobilise (cyclophosphamide à forte dose). Cela permet de constituer un greffon enrichi de ces cellules, pour pallier les effets de la chimiothérapie intensive qui aura lieu quelques semaines plus tard.
Celle-ci consiste en une perfusion de melphalan à forte dose, suivie 24 à 48 heures plus tard de la réinjection de tout ou partie du greffon recueilli. C’est que l’on appelle d’ordinaire une « autogreffe » ou greffe autologue mais qui n’est en réalité qu’un traitement de support permettant de limiter la durée de l’aplasie chimio-induite.
S’ensuit une phase de consolidation, quasi systématique en France,qui reprend la même combinaison qu’à l’induction, en général pour 2 cycles.
Avec cette stratégie, on obtient de très bons taux de réponse et des survies médianes sans progression de l’ordre de 5 à 6 ans.
Le traitement de maintenance par immunomodulateurs (lénalidomide) a reçu l’approbation des autorités de santé. En France, la recommandation est de l’administrer pendant une durée fixe de 2 ans.
Pour les malades âgés
Traitement des rechutes
Perspectives thérapeutiques
Une nouvelle classe, celle des anticorps monoclo- naux anti-CD38, marqueur exprimé à la surface des plasmocytes, fait l’objet d’un programme de développement intensif.9 Parmi ceux-ci, le daratumumab (Darzalex, solution pour perfusion) est aujourd’hui commercialisé pour traiter les rechutes avancées. La tolérance en est très bonne, et une forme sous-cutanée est aujourd’hui en développement. S’il fallait encore des preuves du dynamisme de la recherche clinique dans le myélome, on peut ajouter qu’en 2016, 4 nouvelles molécules ont obtenu leur AMM en 2e ou 3e ligne de traitement.
Traitement d’urgence
Correction des facteurs précipitants : perfusion de bisphosphonates contre une hypercalcémie, arrêt des AINS et des diurétiques de l’anse (ils acidifient les urines), traitement d’une infection intercurrente.
Compression médullaire : sauf syndrome déficitaire objectif et brutal nécessitant une chirurgie de décompression en urgence, la stratégie est de débuter rapidement une corticothérapie à forte dose, en prévoyant sans délai l’irradiation de la lésion en cause.
Suivi rapproché
Le déficit immunest souvent aggravé par le traitement antitumoral, notamment à l’initiation de la dexaméthasone. En conséquence, toute fièvre ou début d’infection respiratoire, impose sans délai une antibiothérapie généralement probabiliste. En première intention, l’amoxicilline-acide clavulanique est une bonne option qui cible surtout le pneumocoque et Haemophilus. Il est, bien sûr, hors de question de prescrire de corticoïdes, qui aggraveraient encore l’immunosuppression.
L’hospitalisation doit être facile, en particulier chez les patients fragiles, ou ayant des difficultés respiratoires.
Une attention soigneuse doit être portée à la fonction rénale, notamment dans les formes à chaînes légères, en particulier lors des épisodes infectieux ou de déshydratation. Une absorption régulière de boissons est nécessaire, pour alcaliniser les urines puisque le pH acide favorise la précipitation des chaînes légères. On demande au patient de choisir des eaux alcalines (Vichy) ou on lui prescrit des gélules de bicarbonate de sodium. Les AINS sont formellement contre-indiqués.
Et il faut éviter si possible l’administration de diurétiques de l’anse (qui acidifient les urines), d’un IEC ou d’un ARA 2.
Critères CRAB : 1 seul est requis pour traiter
Calcium level increase (hypercalcémie) : plus de 0,25 mmol/L au-dessus de la limite supérieure de la normale, ou > 2,75 mmol/L.
Renal insufficiency (insuffisance rénale) : créatininémie > 173 µmol/L ou clairance < 40 mL/min.
Anemia (anémie) : hémoglobine à moins de 2 g/dL sous la limite inférieure de la normale, ou < 10 g/dL.
Bone lesions (atteinte osseuse) : lyses ou tassements vertébraux (d’allure parfois banale, bilan IRM ou TDM).
Que dire à vos patients
Le myélome multiple reste une maladie incurable. La rechute est le plus souvent inéluctable, mais les progrès thérapeutiques augmentent le nombre et la durée des rémissions.
L’Intergroupe francophone du myélome (IFM) mène, depuis plus de 20 ans, une recherche très active, avec de nombreux essais cliniques, essentiels à l’avancement des thérapeutiques.
Le site internet de l’AF3M (Association française des malades du myélome multiple) est consultable à l’adresse suivante : www.af3m.org
2. Leung N, Bridoux F, Hutchison CA, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 2012;120:4292-5.
3. Kyle R, Therneau T, Rajkumar V. Prevalence of Monoclonal Gammopathy of undetermined significance. N Engl J Med 2006;354:1362-9.
4. Merlini G, Stone M. Dangerous small B-cell clones. Blood 2006;108:2520-30.
5. Moreau P, San Miguel J, Sonneveld P, et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology 2017;28 (suppl 4):iv52-iv61.
6. Fouquet G, Guidez S, Herbaux C, Demarquette H, Leleu X. Myélome multiple indolent. Rev Med Interne 2014;32:243-9.
7. Hulin C, Belch A, Shustik C, et al. Updated Outcomes and Impact of Age With Lenalidomide and Low-Dose Dexamethasone or Melphalan, Prednisone, and Thalidomide in the Randomized, Phase III FIRST Trial. J Clin Oncol 2016;34:3609-17.
8. Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016; 30:1005-17.
9. Dimopoulos M, Oriol A, Nahi H, et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med 2016;375:1319-31.
 Encadrés
Encadrés