Diagnostiquer une néphropathie glomérulaire.
Connaître les néphropathies glomérulaires les plus fréquentes (syndrome néphrotique à lésions glomérulaires minimes, glomérulopathie extra- membraneuse, néphropathie à dépôts mésangiaux d’IgA, glomérulonéphrites rapidement progressives), leurs étiologies et les principes de leurs traitements.
Syndrome glomérulaire
Définition
La protéinurie glomérulaire est constituée majoritairement d’albumine, voire de protéines de poids moléculaire supérieur à l’albumine (gammaglobulines). Elle est dite « microalbuminurie » (albuminurie entre 30 et 300 mg/24 h ou mg/g de créatininurie sur échantillon) ou « macroprotéinurie » (albuminurie > 300 mg/24 h ou mg/g de créatininurie sur échantillon, et protéinurie > 0,5 g/24 h ou 0,5 g/g de créatininurie sur échantillon), ou néphrotique (protéinurie > 3 g/24 h et albuminémie < 30 g/L). Les termes « microalbuminurie » et « macroprotéinurie » devraient disparaître pour être remplacés respectivement par une albuminurie stades A2 et A3, par opposition à la protéinurie physiologique ou stade A1, définie par une protéinurie < 0,15 g/24 h ou 0,15 g/g créatininurie et une albuminurie < 30 mg/24 h ou 30 mg/g de créatininurie sur échantillon. La définition du syndrome néphrotique, purement biologique, peut être prise en défaut en cas de protéinurie non glomérulaire mais abondante, associée à une dénutrition responsable d’une hypo-albuminémie (en particulier au cours d'un myélome).
Elle peut être soit sélective (constituée de plus de 80 % d’albumine à l’électrophorèse des protéines urinaires [EPU]), soit non sélective (entre 50 et 80 % d’albumine). Quand une protéinurie glomérulaire est non sélective, les autres constituants de cette protéinurie sont majoritairement des gammaglobulines. Ceci doit être distingué d’une protéinurie mixte, glomérulaire et tubulaire, dans les néphropathies évoluées où la protéinurie contient tous les éléments protéiques sanguins.
Les protéines de poids moléculaire important (> 65 kDa) ne franchissent pas la membrane de filtration glomérulaire saine. Les protéines de faible poids moléculaire (< 65 kDa, c’est-à-dire < à l’albumine) franchissent physiologiquement le filtre glomérulaire mais sont ensuite réabsorbées au niveau tubulaire. Ainsi, une anomalie glomérulaire sera responsable de la présence de protéines de poids moléculaire important (≥ albumine) dans les urines. Une anomalie tubulaire quant à elle sera responsable de la présence de protéines de petit poids moléculaire < à l’albumine, car non réabsorbées au niveau tubulaire. L’albumine est la plus petite protéine physiologiquement retenue (non filtrée dans les urines) par le filtre glomérulaire, à la fois du fait de son poids moléculaire de 65 kDa mais aussi du fait de sa charge négative, repoussée par la membrane de filtration glomérulaire elle-même chargée négativement. En réalité, une faible quantité d’albumine passe la barrière de filtration glomérulaire, mais ne déborde pas les capacités physiologiques de réabsorption proximale. Ainsi au début des lésions du filtre glomérulaire, l’albumine est la seule à franchir le filtre. Plus la membrane de filtration glomérulaire est altérée, plus les protéines de poids moléculaire de plus en plus important pourront franchir ce filtre. Ainsi, une protéinurie glomérulaire non sélective témoigne de lésions glomérulaires plus sévères.
L’hématurie est microscopique (> 10 GR/mm3) ou macroscopique (visible à l’œil nu). L’origine glomérulaire est suspectée devant les arguments suivants :
- si elle est microscopique, les globules rouges sont déformés en microscopie optique conventionnelle ou éteints en contraste de phase (probablement du fait de leur passage dans le filtre glomérulaire et le choc osmotique dans l’anse de Henlé) et parfois sous forme de cylindres hématiques ;
- si elle est macroscopique, l’hématurie est totale (sans renforcement initial ni terminal) et sans caillot (par opposition à une hématurie d’origine urologique).
Il faut rechercher des signes extra-rénaux devant un syndrome glomérulaire, en rapport avec la cause (cutanés, articulaires, pulmonaires, cardiaques, neurologiques...) et des signes éventuels du retentissement de la maladie rénale
La présence d’un syndrome glomérulaire doit faire rechercher une néphropathie glomérulaire. Il existe plusieurs façons de s’orienter :
- l’approche syndromique à travers la présentation clinique ;
- l’approche morphologique (ou anatomopathologique = histologique) ;
- l’approche étiologique.
Approche syndromique
Syndrome néphritique aigu
Présentation clinique
Il s’agit d’un tableau brutal et transitoire (s’installant puis régressant typiquement en quelques jours) associant :- protéinurie glomérulaire, rarement néphrotique ;
- hématurie glomérulaire, parfois macroscopique ;
- insuffisance rénale aiguë (IRA) souvent oligurique ;
- œdèmes ;
- ± HTA.
Anatomopathologie
La biopsie rénale n’est pas indispensable chez l’enfant en cas de présentation typique :- contexte post-streptococcique évident (angine non traitée par antibiotique) ;
- délai d’incubation silencieuse entre le début de l’angine et les premiers signes cliniques néphrologiques habituel de 12 à 14 jours ;
- anurie depuis moins de 48 heures, insuffisance rénale depuis moins de 15 jours ;
- fraction C3 du complément basse < 3 mois (activation de la voie alterne plus que classique malgré la présence de complexes immuns avec anticorps antistreptocoque) et protéinurie < 6 mois.
- biologiquement par une activation de la voie alterne du complément ;
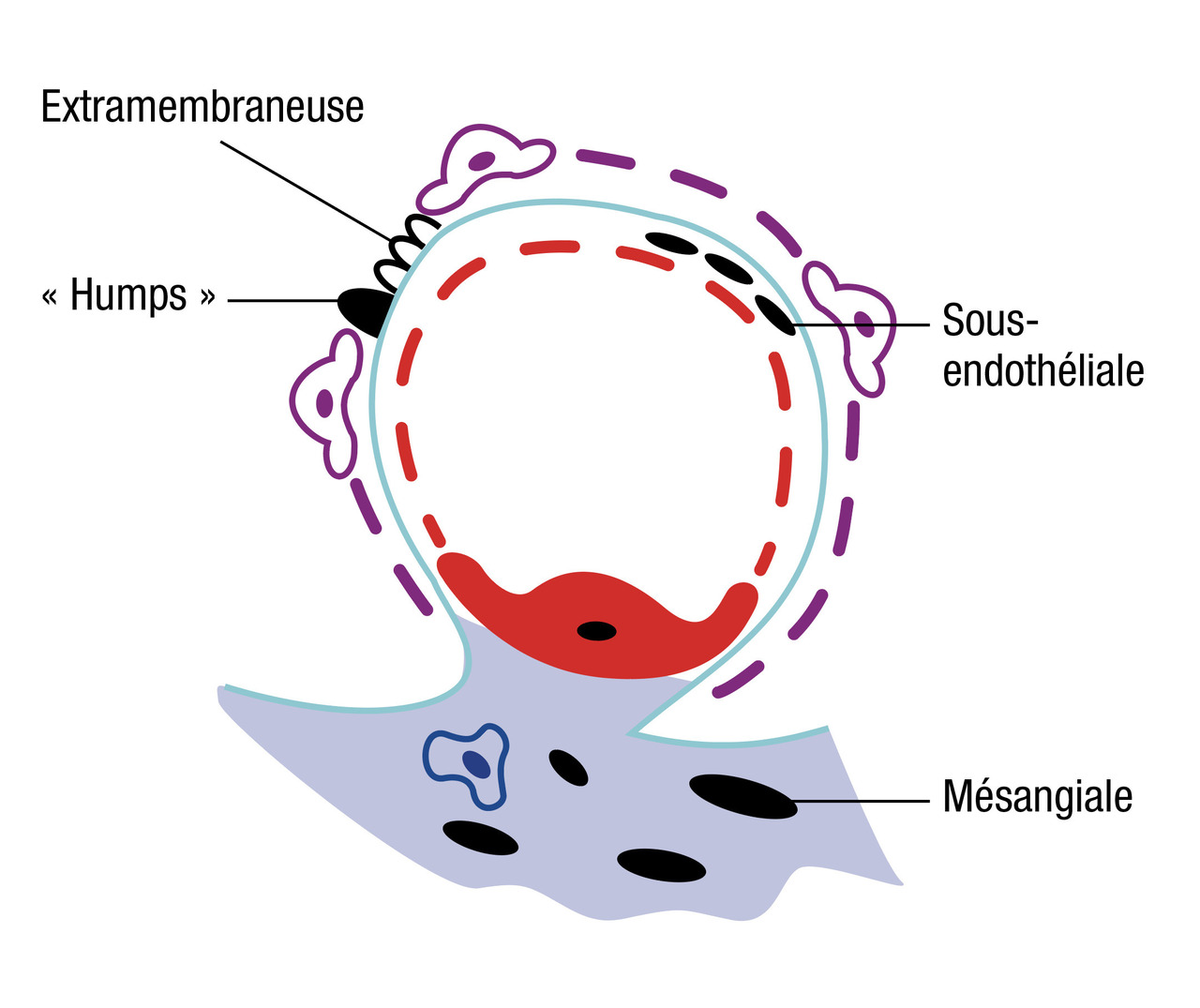
- histologiquement par une prolifération cellulaire endocapillaire diffuse avec parfois quelques dépôts denses en motte sous- épithéliaux (versant externe de la membrane basale) visibles en microscopie optique (humps), et toujours des dépôts diffus en immunofluorescence de fragment de C3 et d'IgG « en ciel étoilé ».
Étiologie
L’agent infectieux est typiquement un streptocoque du groupe A, dans un contexte d’angine. D’autres streptocoques et d’autres localisations sont possibles (abcès dentaire, autre infection ORL, érysipèle…)D’autres agents bactériens ont été incriminés : staphylocoque, pneumocoque…
Traitement
L’hospitalisation est urgente :- pas d’antibiothérapie systématique car l’infection bactérienne causale a typiquement été résolutive ;
- traitement symptomatique (antihypertenseurs, diurétiques, dialyse éventuelle) ;
- surveillance prolongée car réapparition possible de la protéinurie ou de l’insuffisance rénale bien longtemps après l’épisode initial.
- chez l'enfant, insuffisance rénale régressive dans la quasi-totalité des cas, malgré une disparition parfois retardée de la protéinurie, mais risque légèrement plus important de maladie rénale chronique à l’âge adulte ;
- pronostic beaucoup plus réservé chez l’adulte.
Syndrome de glomérulonéphrite rapidement progressive (GNRP)
Présentation clinique
C’est un syndrome glomérulaire sévère associant :- insuffisance rénale rapidement progressive (en quelques semaines) et rarement spontanément régressive (diagnostic différentiel du syndrome néphritique aigu) ;
- hématurie systématique au moins microscopique et parfois macroscopique ;
- protéinurie glomérulaire généralement non néphrotique ;
- souvent sans HTA ni œdèmes initialement.
Anatomopathologie
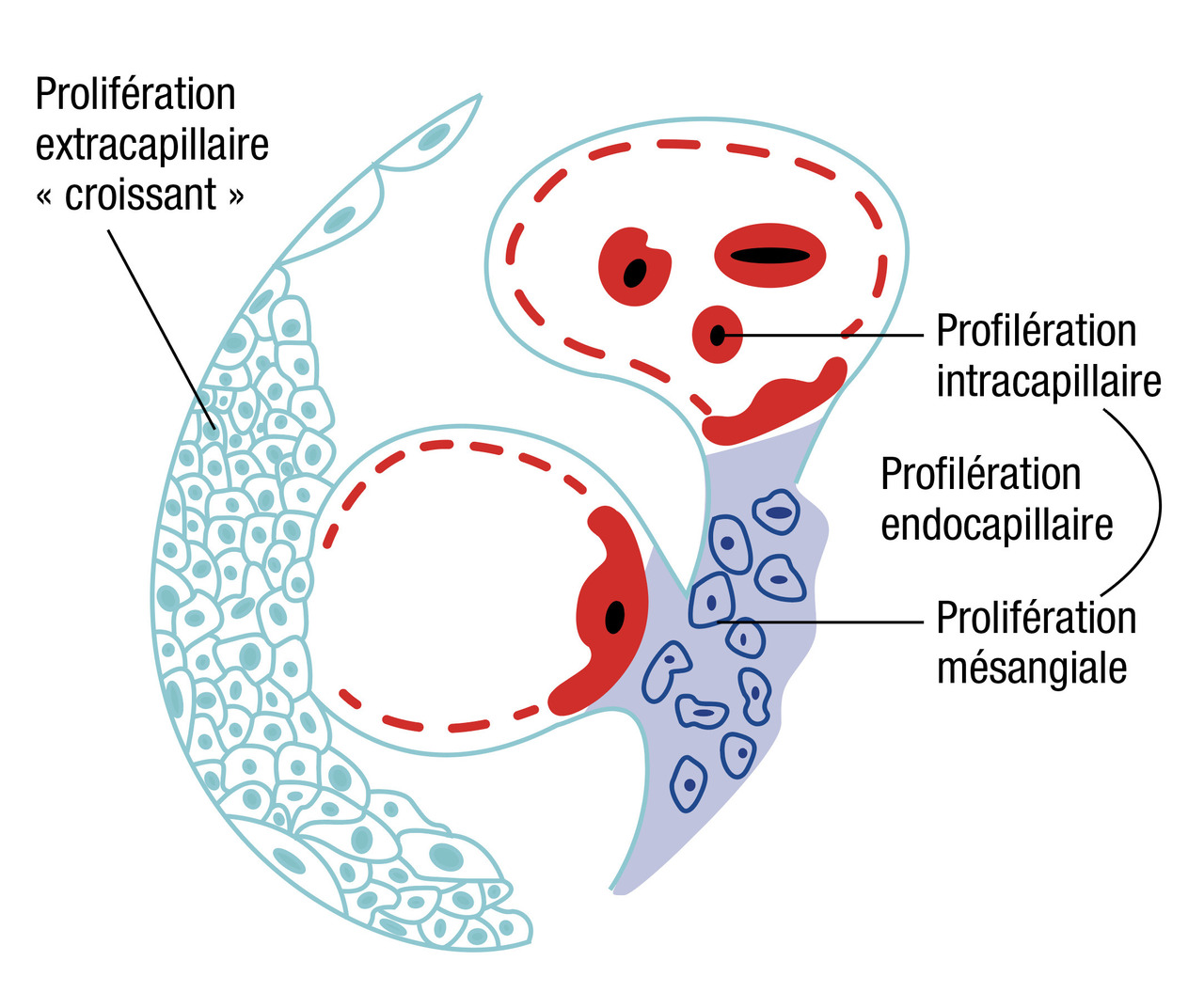
La biopsie rénale est systématique et urgente.Typiquement, elle montre en microscopie optique l’élément commun à toutes les glomérulonéphrites rapidement progressives : la présence de croissants extracapillaires (prolifération des cellules épithéliales dans la chambre urinaire et afflux de cellules inflammatoires)
- l'aspect du floculus : foyers de nécrose dans les vascularites à ANCA (anti-neutrophil cytoplasmatic antibodies), dépôts sous- endothéliaux de cryoglobuline… ;
- la présence de granulomes dans l'interstitium centrés par des vaisseaux dans le cadre de la granulomatose avec polyangéite (ex-Wegener) ou d'une granulomatose éosinophilique avec polyangéite (ex-Churg et Strauss).
Étiologie
Il existe plusieurs types.Type 1. Dépôts linéaires d'IgG le long de la membrane basale glomérulaire (MBG) : maladie de Goodpasture, rare mais sévère :
- anticorps anti-MBG reconnaissant aussi la membrane basale alvéolaire pulmonaire dont la structure moléculaire est identique ;
- expliquant l'association fréquente à une hémorragie intra- alvéolaire avec toux, dyspnée, hémoptysie d'installation parfois dissociée de l’insuffisance rénale aiguë.
- lupus érythémateux aigu disséminé (dépôts d'IgG, IgM, IgA, C3, C4, C1q) ;
- glomérulonéphrite membrano-proliférative : cryoglobulinémie mixte souvent secondaire à une hépatite C (dépôts d'IgG, IgM+C3), post-infectieuse et endocardite (dépôts d'IgA, C3), purpura rhumatoïde (dépôts d'IgA isolés).
- granulomatose avec polyangéite (ex-Wegener) : avec ANCA surtout de type cytoplasmique (en immunofluorescence sur polynucléaires fixés à l’alcool), dirigés contre la protéinase 3 (PR3, enzyme contenue dans les granules primaires intra- cytoplasmiques). Il s'agit d'une une maladie caractérisée cliniquement par une atteinte ORL (rhinite croûteuse, épistaxis, sinusite, otite…), des atteintes nodulaires parfois pseudo-tumorales avec granulomes à cellules géantes sans nécrose caséeuse et une évolution avec rechutes aiguës fréquentes et localisations viscérales multiples ;
- polyangéite microscopique (PAM) : avec ANCA surtout de type périnucléaires dirigés contre la myélopéroxydase (MPO, enzyme contenue dans les granules primaires intracytoplasmiques se localisant autour du noyau du fait de sa charge électrique après fixation des neutrophiles à l’alcool). C'est une maladie définie par défaut en l’absence d’atteinte ORL, nodulaire et de granulome, et évoluant souvent de façon plus insidieuse par poussées successives, avec atteinte rénale parfois isolée ;
- plus rarement, granulomatose éosinophilique avec polyangéite (ex-Churg et Strauss) : surtout MPO-ANCA. C'est une maladie caractérisée par un granulome avec éosinophiles, un asthme grave d'installation récente, et une hyperéosinophilie > 1 500/mm3.
Traitement
L’hospitalisation est urgente.Le traitement se décompose en :
- traitement d'attaque : corticoïdes à fortes doses parfois en bolus IV, cyclophosphamide (Endoxan), et échanges plasmatiques dans la maladie de Goodpasture et les formes sévères de vascularites à ANCA ;
- traitement d'entretien après obtention d’une rémission : corticoïdes et immunosuppresseurs.
Syndrome néphrotique
Présentation clinique
La définition du syndrome néphrotique est purement biologique :- chez l'adulte, protéinurie > 3 g/24 h avec hypo-albuminémie < 30 g/L ;
- chez l’enfant, protéinurie > 50 mg/kg/24 h et hypo-albuminémie < 25-30 g/L.
- œdèmes blancs, mous, indolores, déclives, symétriques, prenant le godet ;
- parfois hypertension artérielle.
- insuffisance rénale aiguë fonctionnelle/chronique organique ;
- thromboses artérielles ou veineuses (en particulier des veines rénales avec majoration du syndrome néphrotique) ;
- surdosage médicamenteux (notamment surdosage en AVK responsable d’hémorragies, et statines avec rhabdomyolyse) ;
- infections à bactéries encapsulées (défaut d’opsonisation par déficit en IgG) : pneumocoque, méningocoque, haemophilus, klebsielle ;
- dyslipidémie : augmentation du LDLc ;
- dénutrition et diminution de la masse musculaire (fuite protéique).
Anatomopathologie
La biopsie rénale est systématique sauf en cas de :- syndrome néphrotique pur chez l'enfant âgé de 1 à 10 ans, car très évocateur de lésion glomérulaire minime ;
- amylose documentée sur une biopsie extrarénale (glande salivaire) ;
- maladie héréditaire connue ;
- diabète ancien mal équilibré avec rétinopathie et en l’absence de signes « atypiques » de néphropathie diabétique (hématurie microscopique principalement).
Étiologie et prise en charge
Le syndrome néphrotique peut être primitif ou secondaire (diabète, amylose...).Le syndrome néphrotique peut être pur ou impur, en cas d’hypertension artérielle associée et/ou d’hématurie et/ou d’insuffisance rénale organique.
Le caractère pur oriente vers une lésion glomérulaire minime (LGM).
Le caractère impur oriente vers tous les autres aspects anatomo-pathologiques :
- hyalinose segmentaire et focale (HSF) ;
- glomérulonéphrite extra-membraneuse (GEM, primitive ou secondaire) ;
- glomérulonéphrite membrano-proliférative (GNMP, primitive ou secondaire) ;
- néphropathie à dépôts mésangiaux d'IgA (primitive ou maladie de Berger limité au rein, ou secondaire avec signes systémiques).
Les causes sont idiopathiques chez l’enfant ou l’adulte jeune ou secondaire : médicaments (AINS ++), lymphome de Hodgkin.
Le traitement spécifique de la forme idiopathique fait appel à la corticothérapie plus prolongée chez l’adulte jeune, avec le traitement adjuvant habituel (restriction sodée et en aliments à haut index glycémique, supplémentation en calcium et vitamine D, apport de potassium adapté à la kaliémie, éventuelle protection gastrique).
Le pronostic est variable selon la corticosensibilité :
- corticosensibilité surtout chez l’enfant, parfois rechutes fréquentes ;
- corticodépendance quand la rechute survient pendant la décroissance ou dans les trois mois suivant l’arrêt des corticoïdes ;
- rares corticorésistance surtout chez l’adulte et en cas de formes génétiques.
Les causes sont idiopathiques chez l’adulte jeune, ou secondaires :
- à une réduction néphronique (rein unique, reflux vésico- urétéral…) ;
- anomalies de l’hémodynamique intrarénale : obésité et drépanocytose ;
- virale : HIVAN (HIV associated nephropathy) surtout chez les sujets noirs ;
- toxique : héroïne IV surtout chez les sujets noirs ;
- maladie héréditaire.
Glomérulonéphrite extramembraneuse : en microscopie optique avec coloration de la membrane basale (argent), on retrouve un aspect épaissi et spiculé de la membrane basale sur son versant externe autour de dépôts granuleux visibles en immunofluorescence d’IgG et de C3 (entre les podocytes).
Les causes sont :
- primitive, associée à des auto-anticorps (anti-PLA2R1 ou plus rarement anti-THSD7A) ;
- secondaires à différentes causes : cancers solides (côlon, poumon, sein…), lupus, infections (HBV++, syphilis, parasitoses comme la lèpre ou les filarioses), médicaments et toxiques (anti-inflammatoires non stéroïdiens [AINS] plus que sels d'or et D-pénicillamine de moins en moins utilisés), sarcoïdose.
- en l’absence de signe d’évolutivité rapide (insuffisance rénale organique), traitement symptomatique pendant au moins 6 mois dans l’espoir d’une rémission spontanée : inhibiteurs du système rénine-angiotensine-aldostérone (SRAA) pour réduire la protéinurie, avec régime hyposodé et diurétiques si œdèmes ; traitement anticoagulant à dose curative si albuminémie < 20 g/L, car les thromboses sont plus fréquentes, en particulier des veines rénales ; statines pour la dyslipidémie.
- en cas de résistance à un traitement symptomatique ou d’évolution vers une insuffisance rénale, un traitement immunosuppresseur peut être discuté en milieu spécialisé.
Type 1 :
- prolifération mésangiale envahissant l’espace sous-endothélial (responsable de l’aspect en double contour de la membrane basale glomérulaire) ;
- dépôts sous-endothéliaux de C3 et IgG ;
- CH50 et C3 souvent abaissés, baisse du C4 inconstante ;
- peut être associée à des causes infectieuses notamment l’hépatite C avec cryoglobulinémie de type 2 (IgM kappa monoclonale avec activité facteur rhumatoïde anti-IgG polyclonale).
- prolifération mésangiale envahissant l’espace sous-endothélial (responsable de l’aspect en double contour de la membrane basale glomérulaire) ;
- dépôts denses de C3 au sein de la membrane basale glomérulaire et tubulaire : aspect rubané ;
- CH50 et C3 abaissés ;
- souvent associée à la présence d’un auto-anticorps activant la voie alterne du complément appelé C3Nef ou « facteur néphritique » ;
- récidive fréquente après transplantation.
- primitive, devenue rare ; le traitement n'est pas clairement codifié ; l’évolution est défavorable ;
- secondaires à la cryoglobulinémie (virus de l'hépatite C) ; infections (VIH, virus de l'hépatite B, virus de l'hépatite C, parasites comme le paludisme, la filariose et l’endocardite) ; lupus ; hémopathies (leucémie lymphoïde chronique et lymphome non hodgkinien) ; carcinomes épidermoïdes.
Syndrome des hématuries macroscopiques récidivantes
Présentation clinique
Comme son nom l’indique, ce syndrome se manifeste par des épisodes d’hématuries macroscopiques récidivantes d’origine non urologique, c’est-à-dire une hématurie totale sans caillot avec des hématies déformées par leur passage dans le filtre glomérulaire si l’on pratiquait un examen au microscope avec contraste de phase.Anatomopathologie
La néphropathie à dépôts mésangiaux d’IgA est la principale cause à évoquer. Elle est caractérisée par des dépôts mésangiaux diffus et nettement prédominants d’IgA, associés à des degrés variables d’épaississement des espaces mésangiaux et de prolifération endocapillaire segmentaire.Cependant, toutes les glomérulonéphrites chroniques peuvent, plus rarement, entraîner une hématurie macroscopique au décours d’un stimulus immunitaire (infection, vaccination).
Étiologie
La néphropathie à IgA peut être primitive ou secondaire.Primitive : maladie de Berger avec une atteinte strictement limitée au rein dont la présentation et l’évolution sont extrêmement variables :
- hématurie microscopique isolée ;
- poussées d’hématuries macroscopiques récidivantes, dès les premiers jours d'une infection souvent virale ORL ;
- hématurie microscopique et protéinurie modérée, d'abord sans puis avec hypertension artérielle et insuffisance rénale progressive ;
- syndrome néphrotique ;
- glomérulonéphrite rapidement progressive.
Traitement
Le traitement spécifique de la maladie de Berger est avant tout symptomatique :- néphroprotection (inhibiteur du système rénine-angiotensine pour le contrôle strict de la pression artérielle et de la protéinurie) ;
- traitement systématique de toute infection ORL ;
- surveillance (pression artérielle, créatininémie, protéinurie) en l’absence de signes de gravité.
Le pronostic est variable : les facteurs de mauvais pronostic sont, outre le sexe masculin, la sévérité de l’hypertension artérielle, de la protéinurie et de l’insuffisance rénale au moment de la prise en charge. Des épisodes isolés d’hématurie macroscopique sont de bon pronostic.
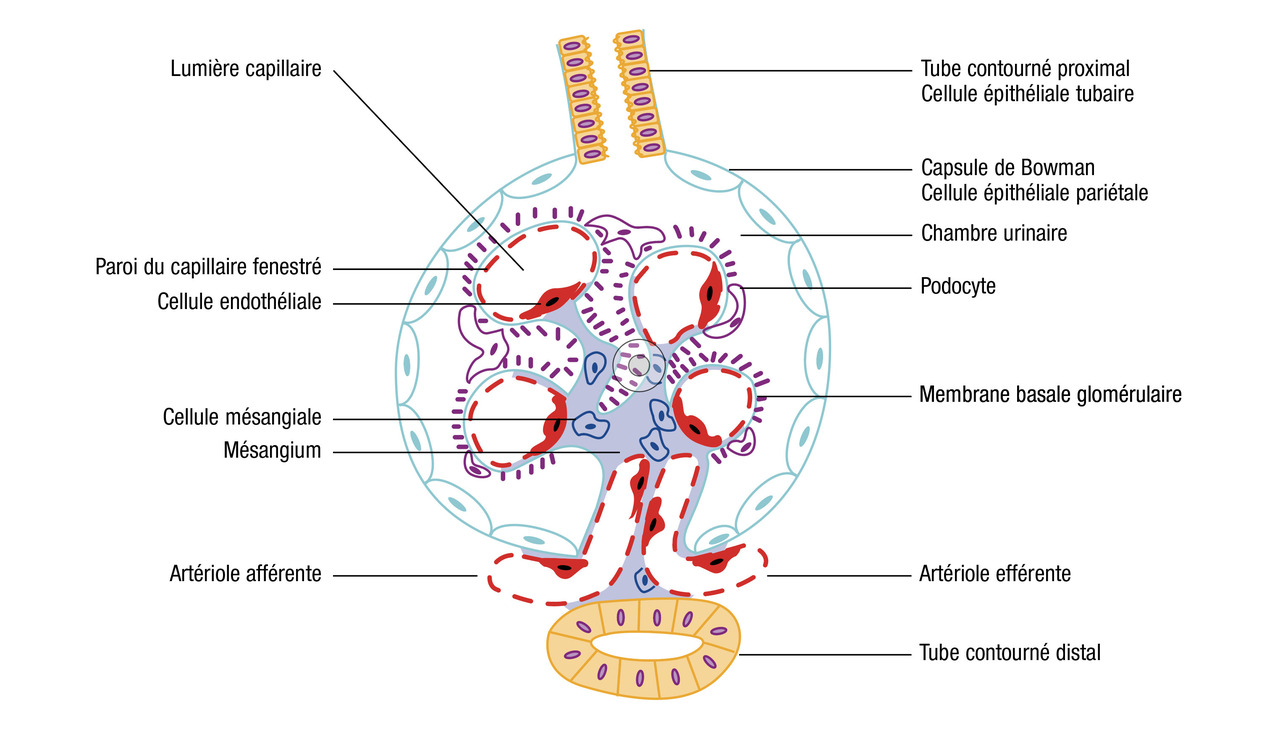
Approche histologique
Anatomopathologie rénale simplifiée
- la microscopie optique (examen au microscope des différentes structures avec différentes colorations)
(fig. 1) ;
- l'immunofluorescence (ayant pour objectif la recherche des dépôts à l’aide d’anticorps spécifiques) ;
- la microscopie électronique quant à elle n’est pas systématique.
Lésions élémentaires des glomérules
Prolifération
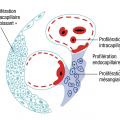
La prolifération correspond à une augmentation du nombre de cellules et se décrit en microscopie optiqueAinsi, la prolifération peut être :
- endocapillaire : il s'agit d'une prolifération des cellules mésangiales et/ou endothéliales c’est-à-dire les cellules du côté du compartiment sanguin ;
- extracapillaire : il s'agit d'une prolifération des cellules du côté du compartiment urinaire, c’est-à-dire des podocytes ou des cellules de la capsule de Bowman. Ces deux types cellulaires sont des cellules épithéliales.
Dépôts
Les dépôts sont documentés par l’examen en immunofluorescencePlusieurs types de dépôts sont possibles :
- immunoglobulines : A, M ou G, chaînes légères (kappa ou lambda)… ;
- fractions du complément : C3, C4, C1q… ;
- facteur de coagulation témoin de lésions actives (fibrine) ;
- substances particulières comme les substances amyloïdes dans l’amylose ;
- protéines des matrices extracellulaires (hyalinose, diabète… ;
- collagène témoin d’une fibrose cicatricielle ;
- lipides : sphingolipides comme dans la maladie de Fabry.
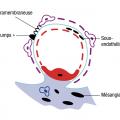
Les dépôts sous-épithéliaux (ou extramembraneux) se situent sous les cellules épithéliales, c'est-à-dire sous les podocytes (entre la membrane basale et les pieds des podocytes).
Les dépôts sous-endothéliaux (ou endomembraneux) se situent sous les cellules endothéliales (entre la membrane basale et les cellules endothéliales de la paroi capillaire).
Les dépôts mésangiaux se trouvent dans le mésangium.
Modifications de la membrane basale
La membrane basale est examinée, à la recherche d’une modification de son aspect global (épaissie, irrégulière, mince…) et d’éventuelles déformations (spicule, chaînette, dédoublement).Nécrose
La nécrose est particulièrement informative lorsqu’elle concerne les anses des capillaires glomérulaires orientant ainsi vers une angéite nécrosante.Distribution des lésions
- par rapport à un glomérule donné : lésions segmentaires si elles ne concernent qu’une portion du glomérule, et lésions globales lorsque les lésions sont présentes sur tout le glomérule ;
- par rapport à l’ensemble des glomérules examinés : lésions focales lorsqu'elles ne concernent qu’un certain pourcentage des glomérules examinés, et lésions diffuses lorsqu’elles concernent tous les glomérules examinés.
Corrélation anatomoclinique et étiologie
- hyalinose segmentaire et focale : protéinurie isolée, hématurie microscopique inconstante et modérée, hypertension artérielle et insuffisance rénale souvent lentement progressive ;
- glomérulonéphrite extramembraneuse : protéinurie isolée souvent néphrotique, hématurie microscopique inconstante et modérée, hypertension artérielle et insuffisance rénale souvent lentement progressive ou rémission spontanée ;
- glomérulonéphrite membrano-proliférative : protéinurie isolée parfois néphrotique, hématurie microscopique, hypertension artérielle et insuffisance rénale parfois rapidement progressive ;
- néphropathie glomérulaire à dépôts mésangiaux d'IgA : hématurie microscopique isolée ou macroscopique récidivante, protéinurie isolée parfois néphrotique ou protéinurie avec hématurie microscopique, hypertension artérielle et insuffisance rénale souvent lentement progressive mais aussi plus rarement rapidement progressive.
Approche étiologique
- les maladies générales : métaboliques, immunologiques… :
. maladies inflammatoires : lupus érythémateux disséminé (LED), purpura rhumatoïde… ;
- les néoplasies solides, les lymphomes et les gammapathies monoclonales malignes (mais aussi bénignes d’un point de vue hématologique mais agressant les reins) ;
- les maladies infectieuses : virus de l'hépatite B, virus de l'hépatite C, VIH, syphilis, germes pyogènes, parasitoses… ;
- les toxiques et médicaments ;
- les maladies familiales et héréditaires.
Maladies générales non inflammatoires
Néphropathie diabétique
La néphropathie diabétique doit être évoquée en cas de micro-albuminurie ou de protéinurie d’origine glomérulaire chez un patient diabétique.La biopsie rénale n’est pas systématique mais doit être réalisée en présence d’éléments cliniques ou biologiques atypiques faisant suspecter une autre origine :
- durée d’évolution insuffisante du diabète (inférieure à 5 ans, surtout dans le cas d’un diabète de type 1, car l’ancienneté est souvent plus difficile à déterminer dans le diabète de type 2) ;
- absence de rétinopathie diabétique (témoin d’une atteinte microvasculaire du diabète) ;
- hématurie microscopique ;
- syndrome néphrotique explosif ;
- insuffisance rénale rapidement progressive ;
- signes en faveur d'une autre maladie générale.
À cet aspect typique peuvent se surajouter des lésions de néphro-angiosclérose liée à l’HTA ancienne chez le patient diabétique de type 2 souvent polyvasculaire hypertendu.
Il n’existe pas de dépôts particuliers en immunofluorescence.
La prise en charge doit être globale :
- équilibrer le diabète : cibles d'HbA1c < 7 % au stade III et < 8 % aux stades IV et V de la maladie rénale chronique ;
- contrôler la microalbuminurie/protéinurie par un inhibiteur du système rénine-angiotensine ;
- traiter l’ensemble des autres facteurs de risque cardiovasculaires ;
- prendre en charge les complications de la maladie rénale chronique.
Amylose
Il s’agit classiquement d’un syndrome néphrotique sans hématurie avec une insuffisance rénale évoluant progressivement. À l’échographie rénale, les reins sont souvent augmentés de volume.Sur le plan clinique, il faut toujours penser à rechercher les atteintes extrarénales : insuffisance cardiaque conditionnant le pronostic vital (surtout amylose AL), syndrome du canal carpien, macroglossie, neuropathie périphérique, hépatomégalie…
Le diagnostic histologique d’amylose peut être porté à partir d’une biopsie d’un autre organe potentiellement plus simple d’accès tel que les glandes salivaires, la graisse sous-cutanée abdominale ou le rectum.
Si une ponction-biopsie rénale est réalisée, elle montre :
- microscopie optique : dépôts glomérulaires amyloïdes, c’est-à-dire des dépôts de chaînes bêta-plissées caractéristiques par leur positivité à la coloration rouge congo ;
- immunofluorescence : elle permet de typer l'amylose en utilisant des anticorps spécifiques des différentes protéines amyloïdes connues ;
- microscopie électronique : elle complète cette description en montrant l’aspect fibrillaire caractéristique des dépôts.
- 2 amyloses acquises : AL et AA ;
- 5 amyloses héréditaires « primitives » que nous ne détaillerons pas car beaucoup plus rares : transthyrétine, fibrinogène Aα, apolipoprotéine A1 ou A2, gelsoline, lysozyme.
Histologiquement, on retrouve des dépôts glomérulaires amyloïdes de chaînes légères le plus souvent lambda.
La prise en charge consiste en une chimiothérapie, plus lourde en cas de myélome que de gammapathie de signification indéterminée.
Le pronostic est déterminé par l’atteinte cardiaque (cardiopathie hypertrophique) à rechercher systématiquement par des dosages de BNP, troponine et par une échographie cardiaque. Une insuffisance cardiaque documentée engage le pronostic vital dans les 6 mois.
Les dépôts de chaînes légères kappa sont plus rarement organisés en feuillets amyloïdes, et sont plus souvent désorganisés, rouge congo négatifs, dans les glomérules et les vitrés tubulaires, constituant un autre syndrome glomérulaire, le syndrome de Randall.
L’amylose AA est secondaire à une maladie inflammatoire évoluant depuis plusieurs années : polyarthrite rhumatoïde, spondylarthrite ankylosante, maladie de Crohn, dilatation des bronches, tuberculose, néoplasie, ou fièvre méditerranéenne familiale (FMF). Les atteintes nerveuse périphérique ou cardiaque symptomatiques sont beaucoup plus rares dans ce type d’amylose.
Histologiquement, il s’agit de dépôts glomérulaires amyloïdes d’un peptide issu du clivage de la protéine SAA synthétisée par le foie en cas d’inflammation chronique.
La prise en charge consiste à traiter la maladie causale. Dans le cas particulier de la fièvre méditerranéenne familiale, on y associe de la colchicine.
Maladies générales inflammatoires
Lupus érythémateux disséminé
L’atteinte rénale est en général précoce, dès la première année d’évolution du lupus. Tous les syndromes glomérulaires sont possibles : protéinurie avec souvent hématurie, syndrome néphrotique impur avec hématurie microscopique, glomérulonéphrite avec insuffisance rénale plus ou moins rapidement progressive.Biologiquement, le lupus est confirmé par la positivité des facteurs antinucléaires et des anticorps anti-ADN natifs, et une consommation du complément par la voie classique (C3 et C4 bas).
La microscopie permet de distinguer 6 classes indépendantes les unes des autres, il n’existe pas d’évolution nécessaire d’une classe à l’autre :
- classe I : rein normal en microscopie optique mais dépôts immuns dans le mésangium en immunofluorescence ;
- classe II : prolifération mésangiale avec dépôts mésangiaux ;
- classe III : prolifération endocapillaire (± extracapillaire) focale (≤ 50 % des glomérules atteints) avec dépôts mésangiaux et sous-endothéliaux, et lésions actives (A) ou chroniques fibreuses (C) ;
- classe IV : prolifération endocapillaire (± extracapillaire) diffuse (> 50 % des glomérules atteints) ; segmentaire (S) ou globale (G), avec dépôts mésangiaux et sous-endothéliaux, et lésions actives (A) ou chroniques (C) ;
- classe V : dépôts extramembraneux sans prolifération : lésions de GEM ;
- classe VI : sclérose glomérulaire (> 90 % des glomérules).
Tous les patients lupiques doivent bénéficier en première intention d’un traitement par antipaludéens de synthèse (avec surveillance de l’électrorétinogramme).
Toutes les classes de néphropathie lupique doivent bénéficier d’une prise en charge globale de leur maladie rénale chronique.
Les glomérulonéphrites de classes III et IV sont souvent des urgences thérapeutiques. Leur traitement comprend une phase d’attaque et une phase d’entretien :
- traitement d’attaque : corticothérapie à forte dose, immunosuppresseur type cyclophosphamide (Endoxan®) ;
- traitement d’entretien : corticothérapie à faible dose.
Purpura rhumatoïde
Il s’agit d’une pathologie affectant préférentiellement les enfants, typiquement les jeunes garçons entre 4 et 8 ans, et souvent précédée par une affection des voies aériennes supérieures.Cliniquement, le purpura vasculaire est constant au niveau des membres inférieurs. Il peut s’accompagner de fièvre, d’une polyarthrite ainsi que de douleurs abdominales, voire d’hémorragie digestive.
L’atteinte rénale est associée dans 30 % des cas et se manifeste en général par une hématurie micro- ou macroscopique avec protéinurie glomérulaire en général modérée, rarement néphrotique. La fonction rénale peut être préservée ou l’insuffisance rénale modérée, et une glomérulonéphrite rapidement progressive est rare.
Le diagnostic est posé grâce à l’examen clinique et la biopsie du purpura s’il est toujours présent qui montrerait des lésions de vascularite avec dépôts granuleux d’IgA.
La biopsie rénale n’est pas nécessaire au diagnostic sauf en cas de doute diagnostique avec un diagnostic différentiel en cas de syndrome néphrotique franc ou de tableau de glomérulonéphrite rapidement progressive.
La biopsie rénale a alors un intérêt pronostique en permettant d’évaluer le degré de prolifération extracapillaire (nombre de croissants) qui est un facteur de mauvais pronostic.
La biopsie rénale, si elle était réalisée, montrerait :
- en microscopie optique, une prolifération endocapillaire ± extra- capillaire en cas de forme grave ;
- en immunofluorescence, des dépôts mésangiaux granuleux d'IgA.
Il n’existe pas de consensus pour le traitement.
Sont traitées par corticothérapie les formes avec atteinte grave de certains organes :
- tube digestif : hémorragie digestive, perforation ;
- rein : insuffisance rénale ou syndrome néphrotique ;
- plus rarement en cas d'autres manifestations : testicules, système nerveux…
L’évolution s’effectue par poussées dans 50 % des cas.
Le pronostic rénal est déterminé par le nombre de croissants (prolifération) extracapillaires.
En raison d’atteintes tardives, la surveillance rénale doit être maintenue pendant 2 ans par l’examen clinique avec surveillance de la pression artérielle et bandelette urinaire.
Maladies infectieuses
Virus
Les principaux virus incriminés sont :- HBV : glomérulonéphrite extramembraneuse ;
- HCV : glomérulonéphrite membrano-proliférative associée à la cryoglobulinémie de type 2 ;
- HIV : hyalinose segmentaire et focale particulière appelée HIVAN (HIV associated nephropathy) chez les hommes noirs principalement.
Bactéries
Les principales bactéries sont :- syphilis : glomérulonéphrite extramembraneuse ++ ;
- germes pyogènes sur foyers infectieux profonds : glomérulonéphrite membrano-proliférative ou amylose AA ;
- infections granulomateuses type lèpre ou tuberculose : amylose AA ;
- streptocoque, endocardite d'Osler, infection sur dérivations atrioventriculaires (shunt nephritis) : glomérulonéphrite aiguë.
Parasites
Ce sont :- filariose (glomérulonéphrite extramembraneuse, glomérulonéphrite membrano-proliférative…) ;
- bilharziose (glomérulonéphrite membrano-proliférative, hyalinose segmentaire et focale…) ;
- paludisme (lésion glomérulaire minime ou glomérulonéphrite extramembraneuse)…
Toxiques et médicaments
Ils sont principalement impliqués dans les lésions glomérulaires minimes et la glomérulonéphrite extramembraneuse, mais il faut penser à les évoquer de manière systématique et à les rechercher par l’anamnèse.Néoplasies
les principales néoplasies à rechercher concernent le sein, le côlon et le poumon. Il s’agit le plus fréquemment d’une atteinte rénale par glomérulonéphrite extra- membraneuse. Le bilan à réaliser dans ce contexte pour éliminer une glomérulonéphrite extramembraneuse secondaire à une néoplasie solide est le suivant : radiographie thoracique, mammographie, ± coloscopie ± fibrobronchique/ORL. Le traitement de la néphropathie consiste à réaliser un traitement curatif du cancer.Lymphomes : principalement les lymphomes hodgkiniens associés aux lésions glomérulaires minimes. Cela implique un examen clinique des aires ganglionnaires devant une lésion glomérulaire minime sans examen complémentaire systématique. Le traitement est étiologique par le traitement du lymphome.
Gammapathies monoclonales : le myélome multiple comme les gammapathies de signification indéterminées peuvent être responsables d’une amylose AL par dépôts glomérulaires de chaînes légères le plus souvent lambda ou bien d’un syndrome de Randall en cas de dépôts glomérulaires non amyloïdes (rouge congo négatifs) de chaînes légères le plus souvent kappa.
Il existe également des glomérulonéphrites membrano-prolifératives associées au myélome multiple en cas de cryoglobulinémie de type I (monoclonale) associée.
Maladies familiales et héréditaires
Il faut évoquer ces causes en cas d’atteinte familiale et effectuer une enquête génétique. Nous n’évoquerons ici que les principales néphropathies glomérulaires familiales :- syndrome d'Alport : maladie liée à l’X, avec anomalie du collagène de type IV et irrégularité de la membrane basale en microscopie électronique, aboutissant dans sa forme typique à une insuffisance rénale terminale avec hématurie et surdité de perception totale chez un garçon entre 20 et 30 ans ;
- maladie de Fabry : déficit lié à l’X en alphagalactosidase, provoquant des dépôts de sphingomyéline vasculaires systémiques et dans l’épithélium glomérulaire, avec typiquement chez un jeune garçon : manifestations douloureuses des extrémités, intolérance à l’effort par hyposudation, troubles digestifs, angiokératomes, et sur le plan rénal une protéinurie et une insuffisance rénale progressive ;
- fièvre méditerranéenne familiale (FMF) : autosomique récessive, inflammation chronique associée à une amylose AA, avec poussées de fièvre, arthrite et épanchement des séreuses.
Principes thérapeutiques
Traitement symptomatique
- le traitement de l’HTA et de la protéinurie : inhibiteur du système rénine-angiotensine ;
- les diurétiques en cas de surcharge extracellulaire ;
- la prise en charge de l'insuffisance rénale chronique en cas d’altération de la fonction rénale
(v. item 253) .
Traitement de la cause
Traitement des causes inflammatoires
- la corticothérapie ;
- les immunosuppresseurs, en prêtant attention au risque infectieux :
. échanges plasmatiques : épuration d’anticorps principalement.
Surveillance
Cliniquement
- la pression artérielle ;
- le poids ;
- l’évaluation de la volémie (hyperhydratation ou déshydratation extracellulaire).
Biologiquement
- la fonction rénale, la protéinurie et la présence d'une hématurie principalement ;
- le retentissement de la maladie rénale chronique (v. item 253) ;
- les marqueurs biologiques de la maladie le cas échéant.
Le syndrome glomérulaire se définit par l’association diverse des signes suivants : protéinurie glomérulaire constituée majoritairement d’albumine voire de protéines de poids moléculaire supérieur à l’albumine (gammaglobulines), hématurie micro- ou macroscopique non urologique, œdèmes, hypertension artérielle, insuffisance rénale.
Un même syndrome clinique peut correspondre à différentes lésions histologiques glomérulaires, et une glomérulopathie peut s’exprimer sous la forme de différentes présentations cliniques syndromiques. Par ailleurs, une glomérulopathie peut être primitive ou secondaire à différentes causes.
Le syndrome de glomérulonéphrite rapidement progressive est une urgence diagnostique et thérapeutique.
Le traitement de ces glomérulopathies comporte toujours un versant symptomatique. De nombreuses glomérulopathies nécessitent en plus un traitement immunosuppresseur. Lorsqu’une étiologie spécifique est identifiée, son traitement spécifique doit être mis en place.
 Encadrés
Encadrés