La neuromyélite optique, à bien différencier de la sclérose en plaques, est un syndrome qui regroupe différentes affections inflammatoires du système nerveux central. Elle évolue par poussées et se caractérise par la présence d’anticorps anti-aquaporine 4. Sa prise en charge repose sur l’administration, en urgence, de bolus de corticoïdes intraveineux à fortes doses pour diminuer au maximum le risque de séquelles, avant de débuter un traitement de fond.
La neuromyélite optique (NMO), ou neuromyelitis optica spectrum disorder (NMOSD), regroupe différentes affections inflammatoires du système nerveux central (SNC). Ses caractéristiques physiopathologiques, cliniques, radiologiques et sa prise en charge thérapeutique diffèrent nettement de celles de la sclérose en plaques (SEP), chef de fil des maladies inflammatoires du SNC. La NMOSD a été initialement décrite en 1894 par Eugène Devic et était autrefois connue sous le nom de maladie de Devic. La première description est celle d’une patiente ayant développé sur un mois une névrite optique bilatérale sévère ainsi qu’une myélite transverse responsable d’une tétraplégie et dont l’issue a été fatale. L’autopsie a révélé l’absence d’atteinte de l’encéphale, contrairement à ce qui était observé dans la SEP. Jusqu’à la fin du XXe siècle, la reconnaissance de la maladie de Devic comme une entité à part entière ou comme une forme particulière de SEP, dite optico-spinale, était débattue. La découverte en 2004 des anticorps anti-aquaporine 4 (AQP4), spécifiques de la maladie, a permis de définitivement clore ce débat.1
Les femmes plus fréquemment atteintes
La prévalence mondiale de la NMOSD varie entre 0,52 et 4,4 pour 100 000 patients selon l’origine géographique.2 Les populations asiatiques et africaines ont un risque augmenté de développer la maladie. Contrairement à la SEP, il n’est cependant pas observé de gradient Nord-Sud influant l’incidence. En revanche, il existe un sex-ratio encore plus prononcé que pour la SEP, avec une atteinte plus fréquente des femmes (9/1 pour la NMOSD contre 3/1 pour la SEP). L’âge médian de survenue de la maladie est de 39 ans, mais elle peut se déclarer à tout âge avec parfois des formes sévères chez les patients de plus de 65 ans. L’association avec une autre maladie auto-immune comme le lupus ou la myasthénie n’est pas rare et concerne environ un quart des patients.
Une évolution par poussées avec des séquelles importantes
La NMOSD est, comme la SEP, une maladie évoluant par poussées. Cependant, les poussées de NMOSD se distinguent par leur sévérité, pouvant aller jusqu’au décès en l’absence de traitement rapide et adapté. Contrairement à la SEP, il n’y a pas d’évolution progressive du handicap. Celui-ci est uniquement lié aux séquelles des poussées.
Les atteintes cliniques typiques des poussées sont en rapport avec la distribution des lésions dans le SNC :
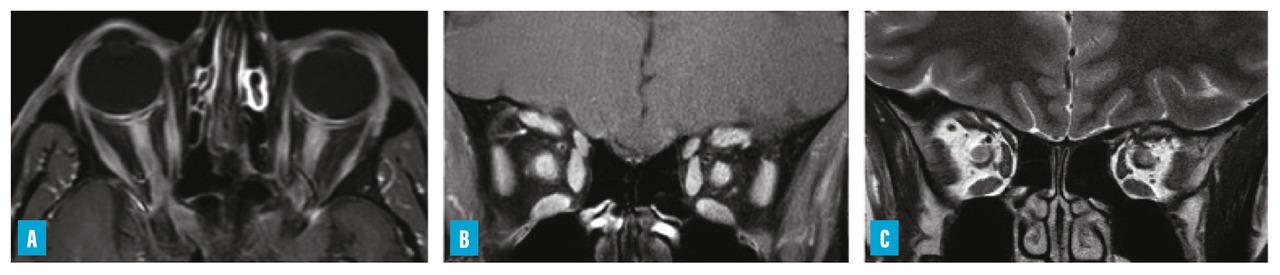
– névrite optique rétrobulbaire (NORB) sévère, volontiers étendue, parfois bilatérale, pouvant toucher le chiasma (
– myélite transverse extensive (étendue sur au moins trois corps vertébraux) avec un syndrome sous-lésionnel sensitif et moteur, et le plus souvent des troubles vésico-sphinctériens ;
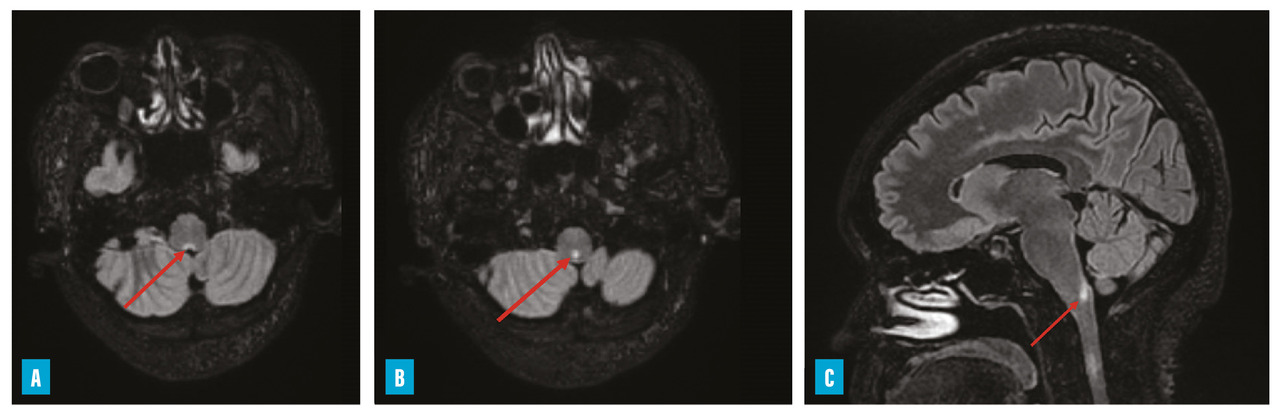
– hoquet et vomissements incoercibles qui peuvent être isolés à la phase initiale (atteinte de la paroi postérieure du 4e ventricule dite area postrema) [
– atteinte des paires crâniennes dans les lésions du tronc cérébral ;
– narcolepsie par atteinte des neurones hypothalamiques sécrétant l’hypocrétine en cas d’atteinte bilatérale diencéphalique ;
– plus rarement, en cas d’atteinte sus-tentorielle, encéphalopathie, voire crises généralisées.
Dans les atteintes les plus sévères, il peut y avoir, à la phase aiguë, une atteinte respiratoire engageant le pronostic vital. La récupération est beaucoup moins bonne qu’après une poussée de SEP, les poussées pouvant laisser des séquelles importantes : tétra- ou paraplégie, cécité, troubles vésico-sphinctériens sévères. Le diagnostic d’une poussée de NMOSD doit donc être rapide, pour une prise en charge thérapeutique en urgence.
Des critères diagnostiques révisés en 2015
Depuis leur première version en 1999,3 les critères diagnostiques ont évolué et ont été révisés en 2010 puis en 2015.4 Actuellement, ils permettent le diagnostic d’une NMOSD si deux atteintes cliniques typiques surviennent de manière distincte dans le temps ; une de ces atteintes au moins doit être une NORB, une myélite longitudinale transverse ou une atteinte de l’area postrema. Par ailleurs, la présence à l’imagerie par résonance magnétique (IRM) de lésions compatibles et en rapport avec les atteintes cliniques est nécessaire, ainsi que l’exclusion d’éventuels diagnostics différentiels. Enfin, si les anticorps anti-AQP4 sont positifs, la présence d’une seule atteinte clinique typique est requise (
La présence d’anticorps anti-aquaporine 4 entraîne une astrocytopathie auto-immune
Les anticorps anti-AQP4 sont des anticorps dirigés contre une protéine des pieds astrocytaires, au contact du liquide céphalorachidien (LCR) : l’aquaporine 4 (AQP4).1 Ceci explique les localisations habituelles des lésions retrouvées dans la maladie : le nerf optique, la moelle épinière, l’area postrema, le tronc cérébral, le diencéphale. Les anticorps anti-AQP4, une fois fixés sur leur cible, déclenchent l’activation de la cascade du complément, entraînant une destruction astrocytaire, et secondairement une démyélinisation et une mort neuronale.5 La physiopathologie est donc bien distincte de celle de la SEP. Ainsi, si la myéline est la cible primaire de la réaction inflammatoire au cours de la SEP, la NMOSD AQP4+ peut être définie comme une astrocytopathie auto-immune. Les causes de l’origine de la production des anticorps anti-AQP4 et les conditions permettant l’initiation du processus lésionnel restent inconnues.
Pour ne pas conclure à tort à une négativité des anticorps anti-AQP4, il est important d’utiliser la technique offrant la meilleure sensibilité et spécificité : la technique de référence est un test cellulaire (cell-based assay [CBA]).6
Éliminer les diagnostics différentiels avec la ponction lombaire
Par ailleurs, la ponction lombaire est indispensable pour ne pas méconnaître un diagnostic différentiel, notamment infectieux. Habituellement, il est constaté une méningite lymphocytaire qui, contrairement à la SEP, peut se manifester par un important taux de leucocytes, jusqu’à plusieurs centaines par millilitre. De plus, la présence de bandes oligoclonales (BOC) est bien moins fréquente que dans la SEP (de 15 à 30 % versus 90 %) ; leur absence constitue un argument supplémentaire pour le diagnostic de NMOSD.7
Différencier NMO et pathologies liées aux anticorps anti-MOG
L’étude du sérum de patients ayant une maladie remplissant les critères de NMOSD mais sans anticorps anti-AQP4 a permis la découverte d’une nouvelle entité caractérisée par la présence d’anticorps anti-MOG. La protéine MOG (myelin oligodendrocyte glycoprotein) est une protéine constitutive de la myéline présente sur la face externe de sa gaine. Les anticorps anti-MOG ont autrefois été décrits dans de nombreuses maladies neurologiques incluant des patients atteints de SEP, voire des sujets sains. Mais des travaux récents ont permis de montrer que, comme pour les anticorps anti-AQP4, la spécificité était bien meilleure en recourant à une technique de détection par CBA permettant d’utiliser la MOG dans sa conformation native.8 Cela a permis la description, en 2011, des premiers cas de patients adultes présentant un phénotype similaire à la NMO, puis aux premières séries de cas en 2014.9, 10 Deux études rétrospectives portant sur des patients ayant présenté une myélite transverse longitudinale étendue mais sans anticorps anti-AQP4 mettaient en évidence des anticorps anti-MOG positifs chez 16 à 23 % des patients.11, 12 Les dernières données montrent cependant que le spectre phénotypique est bien plus étendu : encéphalomyélites aiguës disséminées (ADEM : acute demyelinating encephalomyelitis) de l’enfant, atteintes du tronc cérébral, myélites courtes « SEP-like », encéphalites… Il est maintenant établi qu’il s’agit d’une entité à part entière récemment dénommée MOGAD (MOG antibody-associated disease).
Comme dans la SEP, la physiopathologie des poussées se caractérise par une démyélinisation, mais le profil immunologique des lésions diffère. Par ailleurs, le rôle pathogène direct des anticorps anti-MOG reste à prouver.
Sur le plan épidémiologique, il ne semble pas y avoir de différence de prévalence selon le sexe ou l’origine géographique. La maladie prédomine chez les enfants, avec davantage de formes monophasiques.
Chez les adultes, la névrite optique constitue le mode d’entrée majoritaire dans la maladie (61 % dans la cohorte française MOGADOR).13 Elle est volontiers, comme dans la NMO, bilatérale et sévère. Un œdème papillaire est possible et le segment antérieur du nerf optique est préférentiellement touché. Contrairement à la NMO, l’atteinte du chiasma est rare et la récupération est meilleure. Concernant les myélites, l’atteinte du cône terminal est plus fréquente et doit faire évoquer le diagnostic (
Sur le plan diagnostique, au-delà des caractéristiques cliniques sus-citées, la détection des anticorps dans le sérum est indispensable. Cependant, 13 % des patients ont des anticorps présents seulement dans le liquide céphalorachidien ; leur dosage au cours d’une ponction lombaire est donc indispensable en cas de conviction clinique forte, contrairement à la NMOSD pour laquelle un dosage sérique suffit.
Le pronostic est globalement meilleur que chez les patients présentant des anticorps anti-AQP4, avec une meilleure récupération à distance des poussées. Cependant, en cas de myélite, des séquelles sont possibles, notamment sur le plan vésico-sphinctérien en corrélation avec la plus grande prépondérance des lésions thoraciques. Il existe des formes monophasiques qui sont plus fréquentes chez les patients avec une disparition des anticorps à distance de la poussée.
Prendre en charge les poussées en urgence, puis éviter les récidives
Pour la prise en charge, il faut distinguer le traitement des poussées et le traitement de fond.
Limiter les séquelles
Le traitement de la poussée repose sur des perfusions de corticostéroïdes à fortes doses par voie intraveineuse (méthylprednisolone), à la posologie de 1 g par jour pendant au minimum trois jours et jusqu’à dix jours si nécessaire. En cas d’atteinte sévère, il ne faut pas hésiter à démarrer en parallèle des perfusions, dès J1 ou J2 si besoin, des séances d’échanges plasmatiques qui peuvent être poursuivies le temps de la récupération. La rapidité de l’initiation du traitement conditionne le pronostic de récupération.14
Instaurer un traitement de fond dès la première poussée
Un traitement de fond doit être proposé dès la première poussée de NMOSD et une fois le diagnostic établi. Il vise à empêcher la survenue d’un deuxième événement. Les trois molécules les plus utilisées sont l’azathioprine, le mycophénolate mofétil et le rituximab. Dans une méta-analyse de 2016 portant sur 438 patients traités par rituximab, une diminution moyenne du taux annualisé de poussées de 0,79 (intervalle de confiance à 95 % : -1,08 à -0,49) était constatée.15 Chez la femme en âge de procréer, l’azathioprine est privilégiée par rapport au mycophénolate mofétil, qui est contre-indiqué pendant la grossesse.
Plus récemment, l’éculizumab, un anticorps monoclonal ciblant la protéine C5 du complément empêchant la formation de son complexe terminal, a reçu une autorisation de mise sur le marché (AMM) dans le traitement de la NMOSD. Il est indiqué en cas d’échec de deux lignes de traitement précédentes.
D’autres molécules seront bientôt disponibles en traitement de fond dans la NMOSD :
– le satralizumab, anticorps monoclonal ciblant le récepteur de l’interleukine 6 (IL-6) ;
– l’inébilizumab, anticorps anti-CD19 ciblant le lymphocyte B ;
– le ravulizumab, autre anticorps monoclonal anti-C5 ayant l’avantage de permettre des perfusions toutes les huit semaines au lieu de quatre pour l’éculizumab.
Pas de traitement de fond validé pour la pathologie liée aux anticorps anti-MOG
Pour la pathologie liée aux anticorps anti-MOG, la prise en charge des poussées est identique. Les symptômes sont très corticosensibles et cela peut constituer un argument diagnostique.
En revanche, il n’y a pas de recommandations officielles ni d’essais randomisés concernant le traitement de fond, et les données manquent sur l’efficacité des différents traitements. Il est maintenant établi qu’il ne doit être démarré qu’en cas de pathologie chronique, c’est-à-dire après un deuxième épisode. Par analogie, l’azathioprine, le mycophénolate mofétil et le rituximab sont les molécules les plus utilisées ; cependant, le rituximab semble moins efficace que chez les patients porteurs d’anticorps anti-AQP4. La réponse de plusieurs patients à des cures régulières d’immunoglobulines intraveineuses (Ig IV) a aussi été décrite. Enfin, le satralizumab et des anticorps monoclonaux anti-MOG sont en cours d’évaluation.
Les poussées de NMOSD sont des urgences neurologiques
La NMOSD, définie par des critères cliniques, biologiques et radiologiques en 2015, est un syndrome qui regroupe des affections inflammatoires rares du SNC. Cliniquement, elle se distingue de la SEP principalement par la sévérité des poussées et l’atteinte opticomédullaire prédominante. Les poussées de NMOSD sont des urgences neurologiques qui peuvent engager le pronostic fonctionnel et vital. Un traitement associant bolus de corticoïdes et échanges plasmatiques est indispensable dès le début des symptômes pour diminuer au maximum le risque de séquelles. Le traitement de fond, adapté au cas par cas, est indispensable pour prévenir le risque de rechute ; sa mise en place impose un suivi en centre de référence.
1. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF Fujihara K et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet Lond Engl 2004;364:2106-12.
2. Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, Kleiter I, Chitnis T; GJCF International Clinical Consortium & Biorepository for Neuromyelitis Optica. Demographic and clinical features of neuromyelitis optica: A review. Mult Scler J 2015;21:845-53.
3. Wingerchuk DM, HogancampWF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999;53:1107-14.
4. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177-89.
5. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007;69:2221-31.
6. Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM. Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol 2014;5:290-303.
7. Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J Neurol Sci 2011;306:82-90.
8. Cobo-Calvo Á, Ruiz A, D’Indy H, Poulat AL, Carneiro M, Philippe N et al. MOG antibody-related disorders: Common features and uncommon presentations. J Neurol 2017;264:1945-55.
9. Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;82:474-81.
10. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: A comparative study. JAMA Neurol 2014;71:276-83.
11. Jitprapaikulsan J, Chen JJ, Flanagan EP, Tobin WO, Fryer JP, Weinshenker BG et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein autoantibody status predict outcome of recurrent optic neuritis. Ophthalmology 2018;125:1628-37.
12. Cobo-Calvo Á, Sepúlveda M, Bernard-Valnet R, Ruiz A, Brassat D, Martínez-Yélamos S, et al. Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: Clinical and prognostic implications. Mult Scler Houndmills Basingstoke Engl 2016;22:312-9.
13. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study. Neurology 2018;90: e1858-e1869.
14. Demuth S, Guillaume M, Bourre B, Ciron J, Zephir H, Sirejacob Y, et al. Treatment regimens for neuromyelitis optica spectrum disorder attacks: A retrospective cohort study. J Neuroinflammation 2022;19(1):62.
15. Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: A systematic review and meta-analysis. JAMA Neurol 2016; 73(11):1342-8.
Dans cet article
- Les femmes plus fréquemment atteintes
- Une évolution par poussées avec des séquelles importantes
- Des critères diagnostiques révisés en 2015
- Différencier NMO et pathologies liées aux anticorps anti-MOG
- Prendre en charge les poussées en urgence, puis éviter les récidives
- Les poussées de NMOSD sont des urgences neurologiques