Les anticorps dirigés contre CTLA-4 (Cytotoxic T-Lymphocyte-associated Antigen 4 ; ipilimumab, Yervoy), PD-1 (Programmed Death-1 ; nivolumab, Opdivo ; pembrolizumab, Keytruda) et PD-L1 (Programmed Death-Ligand 1 ; atézolizumab, Tecentriq ; durvalumab, Imfinzi ; avélumab, Bavencio) appartiennent à une nouvelle classe très prometteuse d’anticancéreux baptisée « immunothérapie ».
Ces molécules ont récemment obtenu l’AMM dans le mélanome avancé (non résécable ou métastatique), le cancer bronchique non à petites cellules avancé, le lymphome de Hodgkin en rechute et le carcinome urothélial avancé en rechute ; elles sont également en cours d’essai clinique dans d’autres cancers.
CTLA-4, PD-1 et PD-L1 sont des « checkpoints » impliqués dans le contrôle du système immunitaire. Via ces mécanismes de régulation, les tumeurs inactivent les cellules immunitaires et plus particulièrement les lymphocytes T. Les agents visant ces cibles, appelés Immune Checkpoint Inhibitors (ICI), réactivent donc la réponse immune contre les cellules cancéreuses. Cette action n’est toutefois pas limitée aux lymphocytes spécifiques de la tumeur. Ainsi, des effets secondaires dysimmunitaires sont possibles, pouvant toucher tous les organes. Le mécanisme physiopathologique impliqué est vraisemblablement une réaction auto-immune médiée par les cellules T, comme le confirment les infiltrats lymphocytaires détectés lors des analyses histologiques ou cytologiques après biopsie ou autopsie.1
Les complications neurologiques sont rares par rapport aux autres effets indésirables. Leur incidence globale est d’environ 4 % pour les anticorps anti- CTLA-4, 6 % pour les anti-PD-1 et 12 % pour la combinaison des deux thérapeutiques. Celles dites sévères (grade 3 ou 4) sont peu fréquentes : < 1 % pour tous les types d’ICI.1 Toutefois, compte tenu du risque vital associé, il faut les reconnaître afin d’adresser le patient en urgence vers un centre spécialisé.
Ces molécules ont récemment obtenu l’AMM dans le mélanome avancé (non résécable ou métastatique), le cancer bronchique non à petites cellules avancé, le lymphome de Hodgkin en rechute et le carcinome urothélial avancé en rechute ; elles sont également en cours d’essai clinique dans d’autres cancers.
CTLA-4, PD-1 et PD-L1 sont des « checkpoints » impliqués dans le contrôle du système immunitaire. Via ces mécanismes de régulation, les tumeurs inactivent les cellules immunitaires et plus particulièrement les lymphocytes T. Les agents visant ces cibles, appelés Immune Checkpoint Inhibitors (ICI), réactivent donc la réponse immune contre les cellules cancéreuses. Cette action n’est toutefois pas limitée aux lymphocytes spécifiques de la tumeur. Ainsi, des effets secondaires dysimmunitaires sont possibles, pouvant toucher tous les organes. Le mécanisme physiopathologique impliqué est vraisemblablement une réaction auto-immune médiée par les cellules T, comme le confirment les infiltrats lymphocytaires détectés lors des analyses histologiques ou cytologiques après biopsie ou autopsie.1
Les complications neurologiques sont rares par rapport aux autres effets indésirables. Leur incidence globale est d’environ 4 % pour les anticorps anti- CTLA-4, 6 % pour les anti-PD-1 et 12 % pour la combinaison des deux thérapeutiques. Celles dites sévères (grade 3 ou 4) sont peu fréquentes : < 1 % pour tous les types d’ICI.1 Toutefois, compte tenu du risque vital associé, il faut les reconnaître afin d’adresser le patient en urgence vers un centre spécialisé.
Clinique : polymorphe
Les symptômes neurologiques surviennent typiquement de manière aiguë ou subaiguë, en environ 6 semaines après le début du traitement par ICI. Très hétérogènes, ils peuvent affecter le système nerveux central ou périphérique.
La céphalée est l’événement indésirable neurologique (nAE) le plus fréquent, indépendamment de la gravité. C’est cependant un symptôme peu spécifique et les formes de grade 3 et 4 pourraient être en réalité des méningites non diagnostiquées.
Ainsi une ponction lombaire s’impose dans tous les cas de céphalées sévères chez un patient traité par immunothérapie.
Parmi les nAEs graves, céphalées, encéphalopathies et méningites dominent dans les essais cliniques prospectifs (21 %, 19 % et 15 % respectivement).
Le système nerveux central peut être touché de diverses façons, réalisant des syndromes hétérogènes : maladies démyélinisantes, vascularites cérébrales, encéphalites limbiques, myélites. Les atteintes du système périphérique sont multiples : syndrome myasthénique, neuropathie focale, polyneuropathie, radiculonévrite (syndrome de Guillain- Barré-like), méningoradiculite.1
La céphalée est l’événement indésirable neurologique (nAE) le plus fréquent, indépendamment de la gravité. C’est cependant un symptôme peu spécifique et les formes de grade 3 et 4 pourraient être en réalité des méningites non diagnostiquées.
Ainsi une ponction lombaire s’impose dans tous les cas de céphalées sévères chez un patient traité par immunothérapie.
Parmi les nAEs graves, céphalées, encéphalopathies et méningites dominent dans les essais cliniques prospectifs (21 %, 19 % et 15 % respectivement).
Le système nerveux central peut être touché de diverses façons, réalisant des syndromes hétérogènes : maladies démyélinisantes, vascularites cérébrales, encéphalites limbiques, myélites. Les atteintes du système périphérique sont multiples : syndrome myasthénique, neuropathie focale, polyneuropathie, radiculonévrite (syndrome de Guillain- Barré-like), méningoradiculite.1
Diagnostic
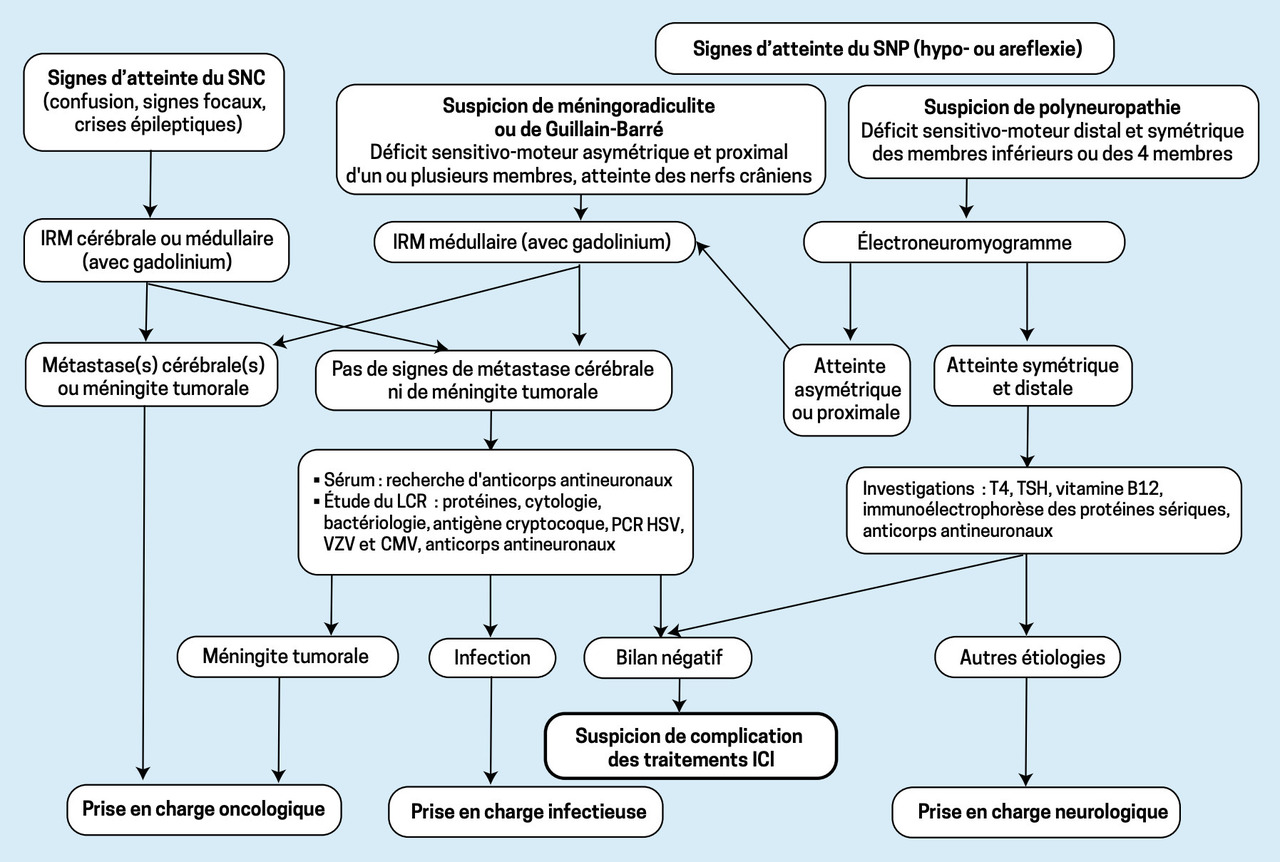
L’apparition de signes neurologiques chez un patient sous ICI doit alerter. Cependant, il faut rechercher systématiquement toutes les autres étiologies, notamment tumorales ou infectieuses.
Le diagnostic de nAEs repose sur 4 paramètres :
– survenue aiguë ou subaiguë ;
– corrélation temporelle entre le début du traitement et celui de la symptomatologie ;
– résultats biologiques (biopsie ou étude du LCR montrant une réaction lymphocytaire) ;
– absence d’arguments pour d’autres étiologies.
En cas de suspicion d’encéphalite, méningite, méningoradiculite ou radiculonévrite (syndrome de Guillain-Barré- like), la ponction lombaire est l’examen le plus utile pour documenter le lien entre signes neurologiques et traitement par ICI. En effet, l’étude du LCR retrouve quasi constamment une hyperprotéinorachie associée à une pléiocytose lymphocytaire (exception faite pour les syndromes de Guillain Barré-like qui se caractérisent par une protéinorachie isolée).
Devant une polyneuropathie, le diagnostic différentiel est plus compliqué. Si la clinique est en faveur d’une atteinte des nerfs périphériques, sans signe radiculaire associé, l’examen du LCR n’est pas indiqué, sauf si l’électroneuro- myographie (ENMG)montre une atteinte démyélinisante et/ou asymétrique, suggérant un Guillain-Barré.
Les syndromes para- néoplasiques ont des caractéristiques physiopathologiques et cliniques similaires aux nAEs, mais dans la grande majorité des cas, on ne retrouve pas les mêmes anticorps anti-neuronaux.2,3,4
Les syndromes myas- théniques liés aux ICI font exception car des anti- corps anti-RACh (récepteur de l’acétylcholine) sont détectés, comme dans la myasthénie classique. Cependant, contrairement à celle-ci, une myosite est souvent associée, avec des douleurs musculaires et une rhabdomyolyse.5
Le diagnostic de nAEs repose sur 4 paramètres :
– survenue aiguë ou subaiguë ;
– corrélation temporelle entre le début du traitement et celui de la symptomatologie ;
– résultats biologiques (biopsie ou étude du LCR montrant une réaction lymphocytaire) ;
– absence d’arguments pour d’autres étiologies.
En cas de suspicion d’encéphalite, méningite, méningoradiculite ou radiculonévrite (syndrome de Guillain-Barré- like), la ponction lombaire est l’examen le plus utile pour documenter le lien entre signes neurologiques et traitement par ICI. En effet, l’étude du LCR retrouve quasi constamment une hyperprotéinorachie associée à une pléiocytose lymphocytaire (exception faite pour les syndromes de Guillain Barré-like qui se caractérisent par une protéinorachie isolée).
Devant une polyneuropathie, le diagnostic différentiel est plus compliqué. Si la clinique est en faveur d’une atteinte des nerfs périphériques, sans signe radiculaire associé, l’examen du LCR n’est pas indiqué, sauf si l’électroneuro- myographie (ENMG)montre une atteinte démyélinisante et/ou asymétrique, suggérant un Guillain-Barré.
Les syndromes para- néoplasiques ont des caractéristiques physiopathologiques et cliniques similaires aux nAEs, mais dans la grande majorité des cas, on ne retrouve pas les mêmes anticorps anti-neuronaux.2,3,4
Les syndromes myas- théniques liés aux ICI font exception car des anti- corps anti-RACh (récepteur de l’acétylcholine) sont détectés, comme dans la myasthénie classique. Cependant, contrairement à celle-ci, une myosite est souvent associée, avec des douleurs musculaires et une rhabdomyolyse.5
Prise en charge thérapeutique
Toute suspicion de complication neurologique sous ICI impose une évaluation spécialisée urgente pour bilan et traitement éventuel. En l’absence d’arguments pour d’autres atteintes, notamment infectieuses, la thérapie par ICI doit être suspendue.
Le traitement n’est pas encore standardisé. En pratique, une corticothérapie orale par prednisone à la dose de 1 mg/kg est instaurée. En raison de la demi-vie lon-gue des anticorps anti-CTLA-4, anti-PD-1 et anti-PDL1, il est préférable de la maintenir au moins 6 semaines, avec un schéma de réduction progressive des doses.
D’autres thérapies telles que les immunosuppresseurs ou la plasmaphérèse n’ont pas fait la preuve de leur efficacité : elles sont réservées aux cas particuliers de résistance aux stéroïdes.
Généralement, la corticothérapie permet une régression rapide des manifestations neurologiques, y compris dans des situations où elle n’est habituellement pas recommandée comme le syndrome de Guillain-Barré. Des décès exceptionnels ont été cependant reportés, principalement secondaires à une détresse respiratoire par atteinte du système nerveux périphérique. Ainsi, prise en charge spécialisée et investigations standardisées sont cruciales afin d’accélérer la gestion et la mise en route du traitement.
La reprise des ICI après un arrêt pour nAE est débattue. Dans notre expérience, elle semble possible en cas de régression complète des symptômes dans des situations sélectionnées : nAE peu sévères (grade 1 et 2), méningite isolée (sans signes d’encéphalite). Une analyse prudente du ratio risque/bénéfice est indispensable au cas par cas.
Le traitement n’est pas encore standardisé. En pratique, une corticothérapie orale par prednisone à la dose de 1 mg/kg est instaurée. En raison de la demi-vie lon-gue des anticorps anti-CTLA-4, anti-PD-1 et anti-PDL1, il est préférable de la maintenir au moins 6 semaines, avec un schéma de réduction progressive des doses.
D’autres thérapies telles que les immunosuppresseurs ou la plasmaphérèse n’ont pas fait la preuve de leur efficacité : elles sont réservées aux cas particuliers de résistance aux stéroïdes.
Généralement, la corticothérapie permet une régression rapide des manifestations neurologiques, y compris dans des situations où elle n’est habituellement pas recommandée comme le syndrome de Guillain-Barré. Des décès exceptionnels ont été cependant reportés, principalement secondaires à une détresse respiratoire par atteinte du système nerveux périphérique. Ainsi, prise en charge spécialisée et investigations standardisées sont cruciales afin d’accélérer la gestion et la mise en route du traitement.
La reprise des ICI après un arrêt pour nAE est débattue. Dans notre expérience, elle semble possible en cas de régression complète des symptômes dans des situations sélectionnées : nAE peu sévères (grade 1 et 2), méningite isolée (sans signes d’encéphalite). Une analyse prudente du ratio risque/bénéfice est indispensable au cas par cas.
références
1. Cuzzubbo S, Javeri F, Tissier M, et al. Neurological adverse events associated with immune checkpoint inhibitors: Review of the literature. Eur J Cancer 2017;73:1-8.
2. Williams TJ, Benavides DR, Patrice KA, et al. Association of Autoimmune Encephalitis With Combined Immune Checkpoint Inhibitor Treatment for Metastatic Cancer. JAMA Neurol 2016;73:928-33.
3. Brown MP, Hissaria P, Hsieh AH, Kneebone C, Vallat W. Autoimmune limbic encephalitis with anti-contactin- associated protein-like 2 antibody secondary to pembrolizumab therapy. J Neuroimmunol 2017;305:16-8.
4. Papadopoulos KP, Romero RS, Gonzalez G, Dix JE, Lowy I, Fury M. Anti-Hu-Associated Autoimmune Limbic Encephalitis in a Patient with PD-1 Inhibitor-Responsive Myxoid Chondrosarcoma. Oncologist 2018;23:118-20.
5. Shirai T, Sano T, Kamijo F, et al. Acetylcholine receptor binding antibody-associated myasthenia gravis and rhabdomyolysis induced by nivolumab in a patient with melanoma. Jpn J Clin Oncol 2016;46:86-8.
2. Williams TJ, Benavides DR, Patrice KA, et al. Association of Autoimmune Encephalitis With Combined Immune Checkpoint Inhibitor Treatment for Metastatic Cancer. JAMA Neurol 2016;73:928-33.
3. Brown MP, Hissaria P, Hsieh AH, Kneebone C, Vallat W. Autoimmune limbic encephalitis with anti-contactin- associated protein-like 2 antibody secondary to pembrolizumab therapy. J Neuroimmunol 2017;305:16-8.
4. Papadopoulos KP, Romero RS, Gonzalez G, Dix JE, Lowy I, Fury M. Anti-Hu-Associated Autoimmune Limbic Encephalitis in a Patient with PD-1 Inhibitor-Responsive Myxoid Chondrosarcoma. Oncologist 2018;23:118-20.
5. Shirai T, Sano T, Kamijo F, et al. Acetylcholine receptor binding antibody-associated myasthenia gravis and rhabdomyolysis induced by nivolumab in a patient with melanoma. Jpn J Clin Oncol 2016;46:86-8.
Dans cet article
essentiel
Les complications neurologiques de l’immuno-thérapie sont rares.
La symptomatologie débute typiquement 6 semaines après l’initiation du traitement.
Toute suspicion impose une évaluation spécialisée en urgence.
La corticothérapie fait régresser les symptômes dans la majorité des cas.