Un enfant sur 10 000 est concerné à la naissance.
Les DSD (développements sexuels différents) regroupent des maladies congénitales responsables d’anomalies du sexe chromosomique, gonadique ou anatomique du nouveau-né.1 Il est parfois impossible de désigner le sexe à la naissance. La découverte d’organes génitaux atypiques est, dans tous les cas, porteuse d’une charge émotionnelle importante, pour les parents et le personnel médical. Il est crucial qu’une information complète soit transmise par des professionnels habitués à ce type de situation.
La prise en charge doit être ciblée et rapide : il faut établir le sexe chromosomique de l’enfant et évaluer le risque vital éventuel d’insuffisance surrénalienne.
La prise en charge doit être ciblée et rapide : il faut établir le sexe chromosomique de l’enfant et évaluer le risque vital éventuel d’insuffisance surrénalienne.
étiologies
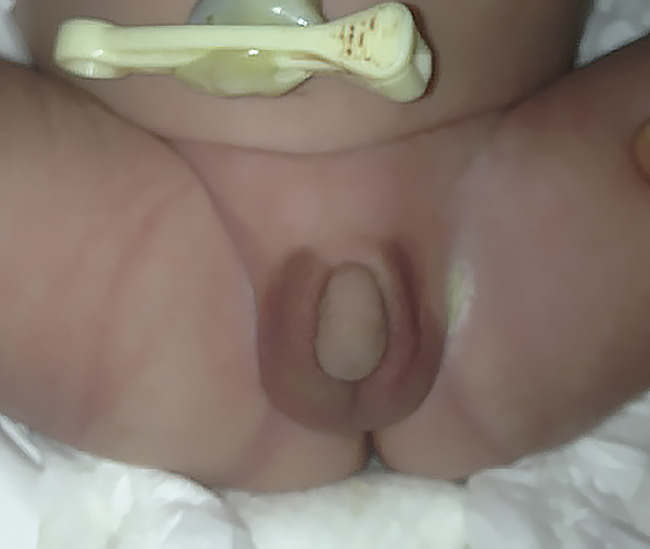
Les seules causes urgentes et graves sont les défauts de la stéroïdogenèse surrénalienne. Le déficit en 21-hydroxylase, le plus fréquent (incidence 1/10 000 à 15 000 naissance en Europe) fait l’objet d’un dépistage néonatal sur papier buvard. Cette maladie autosomique récessive entraîne, chez les filles, une virilisation de degré variable (clitoromégalie ; fig. 1, grandes lèvres fusionnées, orifice unique), alors que chez les garçons il n’y a pas d’anomalie génitale.
Dans les 2 sexes, le risque majeur est celui d’insuffisance surrénalienne, avec dés-hydratation et hypoglycémies. L’élévation de la 17-OH progestérone affirme le diag-nostic. Les autres déficits enzymatiques de la stéroïdogenèse sont beaucoup moins fréquents.
Chez les enfants 46,XX, les autres étiologies sont très rares. L’expression de gènes de la détermination testiculaire, par exemple la translocation du gène SRY sur l’un des X, peut être à l’origine de la virilisation d’un fœtus 46,XX. Une hyper-androgénie maternelle d’origine endogène (lutéomes, tumeur de l’ovaire…) ou exogène (prise de médicaments andro-géniques durant la grossesse) peut également être en cause. Aucune gonade n’est palpée et la virilisation est variable. La cause est toujours retrouvée.
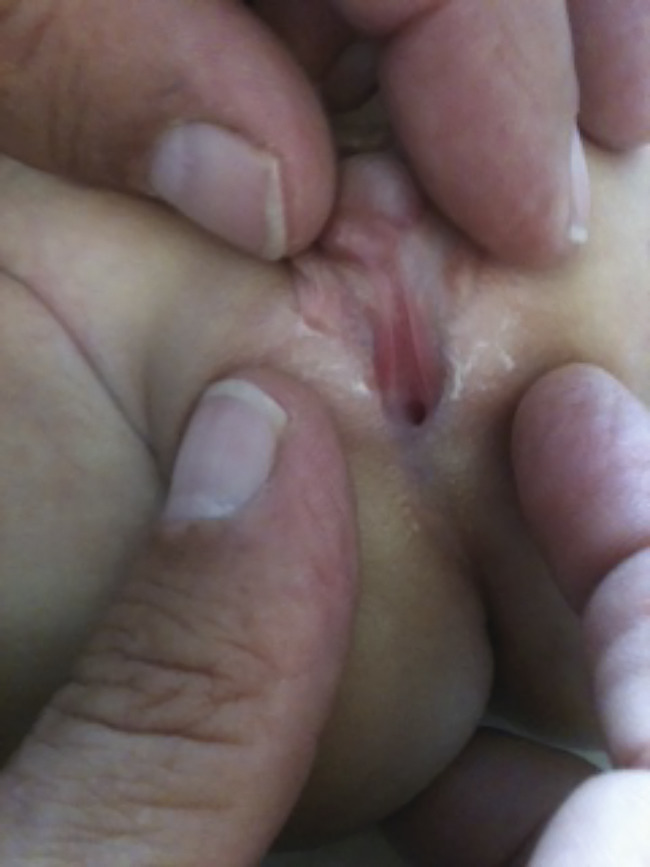
Chez un individu 46,XY avec des organes génitaux atypiques (fig. 2), le diagnostic étiologique n’est possible que dans la moitié des cas environ. La mesure des concentrations de testostérone (T) et d’hormone anti-müllérienne (AMH, produite par le testicule fœtal et responsable de la disparition de l’utérus et du vagin chez les garçons) permet de distinguer les anomalies de la réceptivité à la testo- stérone (AMH normale, pas d’utérus), des dysgénésies testiculaires où les gonades ont sécrété et sécrètent insuffisamment de T et d’AMH (l’utérus n’a pas régressé). Le phénotype est variable : d’un aspect féminin complet à un hypospade périnéal ou scrotal. Les gonades sont situées dans les bourrelets génitaux, en position plus ou moins haute, dans la région inguinale ou en position abdominale.
Des origines non hormonales existent :des anomalies du développement génital sont observées dans de nombreux syndromes malformatifs. L’hypospade est plus fréquent chez les nourrissons nés petits pour l’âge gestationnel.
Dans les 2 sexes, le risque majeur est celui d’insuffisance surrénalienne, avec dés-hydratation et hypoglycémies. L’élévation de la 17-OH progestérone affirme le diag-nostic. Les autres déficits enzymatiques de la stéroïdogenèse sont beaucoup moins fréquents.
Chez les enfants 46,XX, les autres étiologies sont très rares. L’expression de gènes de la détermination testiculaire, par exemple la translocation du gène SRY sur l’un des X, peut être à l’origine de la virilisation d’un fœtus 46,XX. Une hyper-androgénie maternelle d’origine endogène (lutéomes, tumeur de l’ovaire…) ou exogène (prise de médicaments andro-géniques durant la grossesse) peut également être en cause. Aucune gonade n’est palpée et la virilisation est variable. La cause est toujours retrouvée.
Chez un individu 46,XY avec des organes génitaux atypiques (fig. 2), le diagnostic étiologique n’est possible que dans la moitié des cas environ. La mesure des concentrations de testostérone (T) et d’hormone anti-müllérienne (AMH, produite par le testicule fœtal et responsable de la disparition de l’utérus et du vagin chez les garçons) permet de distinguer les anomalies de la réceptivité à la testo- stérone (AMH normale, pas d’utérus), des dysgénésies testiculaires où les gonades ont sécrété et sécrètent insuffisamment de T et d’AMH (l’utérus n’a pas régressé). Le phénotype est variable : d’un aspect féminin complet à un hypospade périnéal ou scrotal. Les gonades sont situées dans les bourrelets génitaux, en position plus ou moins haute, dans la région inguinale ou en position abdominale.
Des origines non hormonales existent :des anomalies du développement génital sont observées dans de nombreux syndromes malformatifs. L’hypospade est plus fréquent chez les nourrissons nés petits pour l’âge gestationnel.
Quand évoquer le diagnostic ?
Le diagnostic est parfois fait en prénatal, lorsqu’un antécédent familial est connu ou en cas de discordance entre l’amniocentèse (sexe chromosomique) et l’échographie. Le plus souvent on découvre l’anomalie à la naissance.
L’observation des organes génitaux fait partie de l’examen clinique du nouveau-né. Des explorations doivent être pratiquées dans les situations suivantes :
– cryptorchidie bilatérale (testicules non palpés) ;
– hypospade postérieur chez un enfant ayant un aspect de garçon ;
– hypospade (même paraissant banal), si la verge est courte et/ou s’il existe une cryptorchidie ou un scrotum bifide ;
– aspect « inclassable » des organes génitaux ;
– orifice vaginal non visible ou fusion postérieure des bourrelets génitaux chez un enfant ayant un aspect féminin ;
– hypertrophie isolée du clitoris ;
– organes génitaux externes féminins avec une masse uni- ou bilatérale dans les grandes lèvres ou les canaux inguinaux, faisant suspecter la présence de testicules.
L’observation des organes génitaux fait partie de l’examen clinique du nouveau-né. Des explorations doivent être pratiquées dans les situations suivantes :
– cryptorchidie bilatérale (testicules non palpés) ;
– hypospade postérieur chez un enfant ayant un aspect de garçon ;
– hypospade (même paraissant banal), si la verge est courte et/ou s’il existe une cryptorchidie ou un scrotum bifide ;
– aspect « inclassable » des organes génitaux ;
– orifice vaginal non visible ou fusion postérieure des bourrelets génitaux chez un enfant ayant un aspect féminin ;
– hypertrophie isolée du clitoris ;
– organes génitaux externes féminins avec une masse uni- ou bilatérale dans les grandes lèvres ou les canaux inguinaux, faisant suspecter la présence de testicules.
Quelle démarche ?
Tout d’abord, il faut faire une description anatomique précise. Rarement, certaines anomalies du développement génital font discuter un choix de sexe différent du sexe chromosomique.
D’autres fois, le phénotype paraît très masculin, et l’enfant est pourtant de sexe féminin (hyperplasie congénitale des sur- rénales).
La recherche de gonades palpables dans les bourrelets génitaux ou dans le canal inguinal est primordiale pour le raisonnement étiologique : si une gonade est palpée, c’est a priori un testicule, et la virilisation incomplète d’un nouveau-né au caryotype 46,XY ou 45,X/46,XY est la plus probable. Si aucune gonade n’est palpée, le bébé est peut-être une fille 46,XX virilisée, et une hyperplasie congénitale des surrénales doit être éliminée de façon urgente.
L’examen clinique, complet, recherche une dysmorphie, des anomalies squelettiques, rénales, anales ou cutanées.
Depuis le décret n° 2017-278 du 2 mars 2017, le délai de déclaration de naissance est de 5 jours. Si le sexe ne peut être défini, il est possible de surseoir à la déclaration de l’enfant. L’article 288 du code d’état civil autorise, si le médecin ne peut pas donner d’indication sur le sexe probable du nouveau-né, à demander au procureur de la République qu’aucune mention de sexe ne soit initialement inscrite dans l’acte de naissance. Les parents sont invités à donner à leur enfant un prénom mixte.
D’autres fois, le phénotype paraît très masculin, et l’enfant est pourtant de sexe féminin (hyperplasie congénitale des sur- rénales).
La recherche de gonades palpables dans les bourrelets génitaux ou dans le canal inguinal est primordiale pour le raisonnement étiologique : si une gonade est palpée, c’est a priori un testicule, et la virilisation incomplète d’un nouveau-né au caryotype 46,XY ou 45,X/46,XY est la plus probable. Si aucune gonade n’est palpée, le bébé est peut-être une fille 46,XX virilisée, et une hyperplasie congénitale des surrénales doit être éliminée de façon urgente.
L’examen clinique, complet, recherche une dysmorphie, des anomalies squelettiques, rénales, anales ou cutanées.
Depuis le décret n° 2017-278 du 2 mars 2017, le délai de déclaration de naissance est de 5 jours. Si le sexe ne peut être défini, il est possible de surseoir à la déclaration de l’enfant. L’article 288 du code d’état civil autorise, si le médecin ne peut pas donner d’indication sur le sexe probable du nouveau-né, à demander au procureur de la République qu’aucune mention de sexe ne soit initialement inscrite dans l’acte de naissance. Les parents sont invités à donner à leur enfant un prénom mixte.
Premier bilan
On recherche par FISH ou PCR le gène SRY (présent sur le chromosome Y, induit la différenciation masculine). Le caryotype complet est réalisé dans un second temps.
Le dosage de la 17-OH progestérone est indispensable pour dépister un certain nombre de maladies responsables d’insuffisance surrénalienne engageant le pronostic vital (cf. supra).2
Testostérone et AMH sont à mesurer avant 36 heures de vie.
L’échographie pelvienne recherche un utérus, des gonades ou des malformations associées.
Le dosage de la 17-OH progestérone est indispensable pour dépister un certain nombre de maladies responsables d’insuffisance surrénalienne engageant le pronostic vital (cf. supra).2
Testostérone et AMH sont à mesurer avant 36 heures de vie.
L’échographie pelvienne recherche un utérus, des gonades ou des malformations associées.
Prise en charge
Le transfert rapide de l’enfant dans un centre spécialisé permet un bilan étiologique complet. La prise en charge est loin d’être simple. Les décisions thérapeutiques dépendent de l’anatomie des organes génitaux et des possibilités chirurgicales (cavité vaginale, utérus, taille du bourgeon génital...), du diagnostic étiologique et de l’évolution prévisible, en particulier en période post-pubertaire.
Dans l’hyperplasie congénitale des surrénales, le traitement comprend de l’hydrocortisone (glucocorticoïdes), du Flucortac (minéralocorticoïdes) et du sel.
De nombreuses questions sont soulevées, en particulier concernant le droit à mettre en place un traitement médical et chirurgical sans l’avis de l’intéressé.
Depuis les années 1950, les décisions concernant le choix du sexe et la chirurgie génitale étaient guidées par la politique du choix du sexe « optimal », censé correspondre à une identité de genre stable, une bonne adaptation psychosociale et la possibilité d’une fonction sexuelle « normale ». Cette approche reposait sur un triple postulat : l’identité de genre (c’est-à-dire l’identification de soi-même comme une fille/une femme ou un garçon/un homme) n’est pas établie à la naissance mais résulte surtout de l’éducation (« nature vs nurture ») ; l’apparence des organes génitaux externes doit être en adéquation avec le genre assigné dès le plus jeune âge ; une identité sexuée stable et la capacité d’une sexualité sont des prérequis à une bonne qualité de vie.
Ces pratiques sont questionnées depuis plusieurs années, sous l’impulsion des associations de patients. L’obligation de déclarer le sexe à la naissance est considérée par certains comme une atteinte aux droits de ces individus, au respect de leur identité.
En France, un enfant aux organes génitaux différents, dont la déclaration de sexe est difficile ou impossible à la naissance, devra néanmoins être rattaché à un sexe social, masculin ou féminin, dans lequel ses parents l’élèveront.3
Dans l’hyperplasie congénitale des surrénales, le traitement comprend de l’hydrocortisone (glucocorticoïdes), du Flucortac (minéralocorticoïdes) et du sel.
De nombreuses questions sont soulevées, en particulier concernant le droit à mettre en place un traitement médical et chirurgical sans l’avis de l’intéressé.
Depuis les années 1950, les décisions concernant le choix du sexe et la chirurgie génitale étaient guidées par la politique du choix du sexe « optimal », censé correspondre à une identité de genre stable, une bonne adaptation psychosociale et la possibilité d’une fonction sexuelle « normale ». Cette approche reposait sur un triple postulat : l’identité de genre (c’est-à-dire l’identification de soi-même comme une fille/une femme ou un garçon/un homme) n’est pas établie à la naissance mais résulte surtout de l’éducation (« nature vs nurture ») ; l’apparence des organes génitaux externes doit être en adéquation avec le genre assigné dès le plus jeune âge ; une identité sexuée stable et la capacité d’une sexualité sont des prérequis à une bonne qualité de vie.
Ces pratiques sont questionnées depuis plusieurs années, sous l’impulsion des associations de patients. L’obligation de déclarer le sexe à la naissance est considérée par certains comme une atteinte aux droits de ces individus, au respect de leur identité.
En France, un enfant aux organes génitaux différents, dont la déclaration de sexe est difficile ou impossible à la naissance, devra néanmoins être rattaché à un sexe social, masculin ou féminin, dans lequel ses parents l’élèveront.3
références
1. Hughes IA, Houk C, Ahmed SF, Lee PA; LWPES Consensus Group; ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child 2006;91:554-63.
2. Plotton I, Gay CL, Bertrand AM, et al. AMH determination is essential for the management of 46,XY DSD patients. Horm Res 2009;72(suppl 3):365.
3. Ahmed SF, Achermann JC, Arlt W, et al. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol (Oxf) 2011;75:12-26.
2. Plotton I, Gay CL, Bertrand AM, et al. AMH determination is essential for the management of 46,XY DSD patients. Horm Res 2009;72(suppl 3):365.
3. Ahmed SF, Achermann JC, Arlt W, et al. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol (Oxf) 2011;75:12-26.