Expliquer l’épidémiologie, les facteurs favorisants et l’évolution des principales pathologies auto-immunes d’organes et systémiques.
Interpréter les anomalies biologiques les plus fréquentes observées au cours des pathologies auto-immunes.
Argumenter les principes du traitement et de la surveillance au long cours d’une maladie auto-immune.
Physiopathologies des maladies auto-immunes
Auto-immunité
Auto-immunité et mécanismes lésionnels
- une tolérance centrale (sélection au niveau de la moelle ou du thymus des lymphocytes T et B reconnaissant le soi) ;
- une tolérance périphérique (anergie des lymphocytes, destruction par apoptose, etc.).
Terrain génétique prédisposant
Facteurs environnementaux
Le statut hormonal est également un élément important, ce qui explique la prédisposition féminine de la plupart des maladies auto-immunes, et les risques d’aggravation sous œstrogènes et lors de la grossesse dans certaines maladies auto-immunes, notamment le lupus.
Classification des maladies auto-immunes
- spécifiques d’un organe, du fait de l’existence d’anticorps dirigés contre une protéine présente spécifiquement dans cet organe (par exemple, les anticorps anti-thyroglobuline dirigés contre la thyroglobuline thyroïdienne dans les thyroïdites auto-immunes) [
tableau 1 ] ;
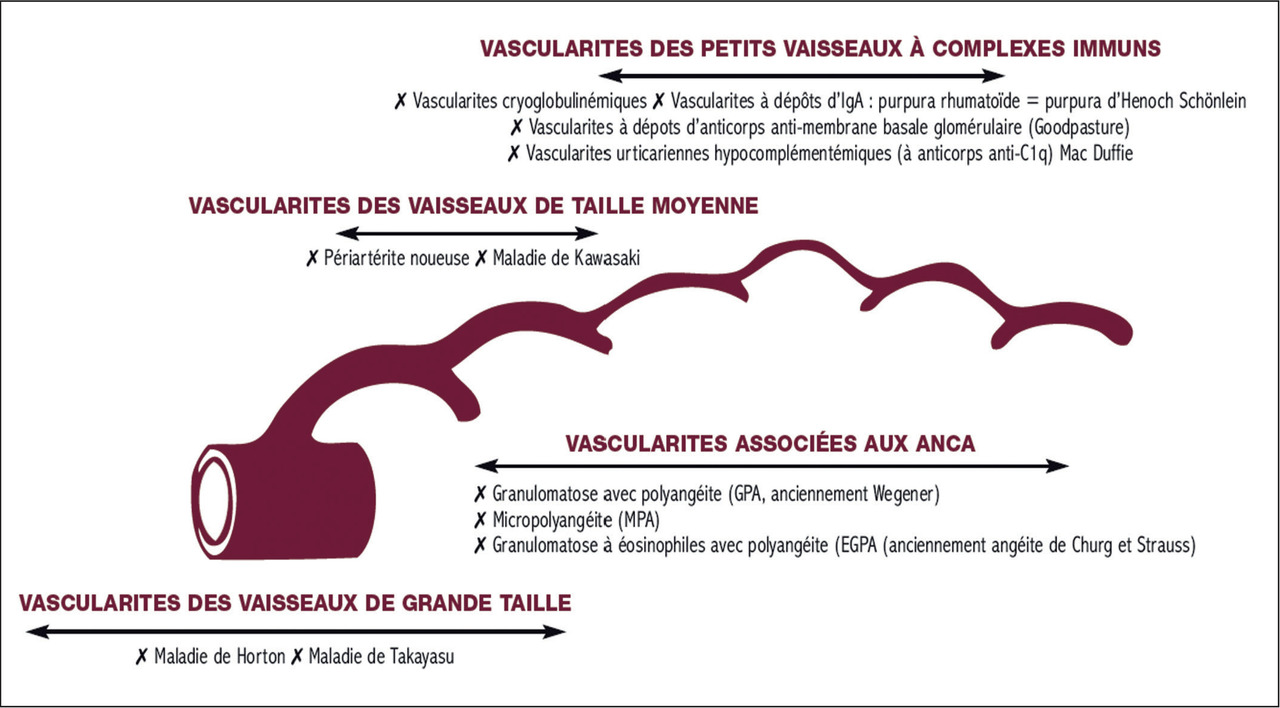
- ou non spécifiques d’organe, à l’origine d’atteintes d’organes différents, du fait de la présence d’autoanticorps de tropisme moins spécifique (par exemple, anticorps antinucléaires, anti- ADN au cours du lupus et anticorps anticytoplasme des polynucléaires neutrophiles dans les vascularites des petits vaisseaux) [
tableau 2 ].
Anomalies biologiques au cours des pathologies auto-immunes
Auto-anticorps
Les auto-anticorps sont des anticorps synthétisés par l’organisme et dirigés contre les auto-antigènes (antigènes du soi), qui sont principalement des acides nucléiques et des protéines. Ces auto-antigènes peuvent être spécifiques ou non spécifiques d’organes dirigés contre un antigène présent dans de nombreux tissus.
La recherche d’un auto-anticorps doit être motivée par un contexte clinico-biologique évocateur d’une maladie auto-immune.
On peut distinguer schématiquement 3 situations amenant à la recherche d’un auto-anticorps.
Un tableau clinico-biologique d’emblée évocateur d’une maladie auto- immune particulière, où la recherche de l’auto-anticorps conforte la suspicion clinique (par exemple, anticorps antinucléaires et anti-ADN chez une femme jeune présentant des arthralgies inflammatoires et un vespertilio, symptômes évocateurs de lupus)
La présence de manifestations systémiques, dont l’ensemble n’oriente pas vers une maladie auto-immune spécifique (par exemple des arthralgies et des myalgies diffuses associées à une altération de l’état général et un syndrome inflammatoire chez un homme d’âge moyen).
La recherche d’un auto-anticorps fait partie de façon systématique du bilan étiologique :
- d'une hypothyroïdie de la femme jeune, où la recherche systématique des anticorps antithyroïdiens (anti-thyroperoxydase [TPO]) permet d’orienter vers une thyroïdite auto-immune ;
- des anomalies des tests hépatiques inexpliqués où la recherche des anticorps anti-mitochondries permet d’évoquer une cirrhose biliaire primitive ;
- des thromboses récidivantes chez le sujet jeune où la recherche systématique des anticorps antiphospholipides permet d’orienter vers un syndrome des antiphospholipides).
- les anticorps peuvent être absents au début de la maladie (ex : polyarthrite rhumatoïde,…),
- ces anticorps ne sont pas constants dans toutes les maladies auto-immunes (seulement 40 % des vascularites à anticorps anticytoplasme des polynucléaires neutrophiles de type granulomatose éosinophile avec polyangeite ont des anticorps anti- cytoplasme des polynucléaires neutrophiles…) ;
- leur détection dépend de la technique utilisée et du seuil de chaque laboratoire
La présence d’un auto-anticorps en l’absence de manifestations cliniques caractéristiques peut parfois précéder de plusieurs mois ou années l’apparition d’une maladie auto-immune. Ainsi, la détection des anticorps antithyroïdiens chez un patient sans signe de dysthyroïdie peut précéder l’apparition d’une hypothyroïdie, il en est de même pour les anticorps antinucléaires précédant l’apparition d’un lupus ou les anticorps antipeptides citrulinés dans la polyarthrite rhumatoïde. Il est habituellement proposer une surveillance clinique régulière et un contrôle régulier des auto-anticorps.
Différents types d’auto-anticorps
Il est important de distinguer 2 types d’auto-anticorps :- les auto-anticorps spécifiques d’organes, que l’on détecte dans le cadre des maladies auto-immunes spécifiques d’organes, comme les anticorps antithyroïdiens dans les thyroïdites auto- immunes ou les anticorps antifacteur intrinsèque dans la maladie de Biermer (tableau 1) ;
- les auto-anticorps non spécifiques d’organes, comme les anticorps antinucléaires (AAN) ou les anticorps anticytoplasme des polynucléaires (ANCA), présents dans les maladies auto- immunes systémiques, comme le lupus ou les vascularites. Les auto-anticorps non-spécifiques d’organes les plus souvent utilisés sont :
- les anticorps antinucléaires ;
- les anticorps antiphospholipides ;
- les anticorps anticytoplasme des polynucléaires.
Anticorps antinucléaires
Deux méthodes sont habituellement utilisées pour rechercher les anticorps anti-nucléaires: l’immunofluorescence indirecte (IFI), et l’enzyme linked immunosorbent assay (ELISA). L’IFI est une méthode très sensible, mais peu spécifique et est utilisée comme test de dépistage. La fluorescence observée en IFI peut avoir différents aspects (homogène, moucheté ou nucléolaire) et les différents aspects de fluorescence correspondent à différentes spécificités (cibles) des anticorps. Par exemple, l’aspect homogène est typiquement associé à la présence d’anticorps anti- ADN (évocateur d’un lupus érythémateux systémique), alors que l’aspect moucheté correspond à la présence d’auto-anticorps anti-antigènes nucléaires solubles (anti-ECT) présents dans plusieurs maladies auto-immunes (Anticorps anticytoplasme des polynucléaires (ANCA)
Les anticorps anticytoplasme des polynucléaires neutrophiles sont dirigés contre des antigènes localisés dans le cytoplasme des polynucléaires neutrophiles. Leur recherche est habituellement motivée par un contexte clinico-biologique (Les anticorps anticytoplasme des polynucléaires neutrophiles sont utiles pour le diagnostic et le suivi des vascularites systémiques à anticorps anticytoplasme des polynucléaires neutrophiles, parmi lesquelles (
- la granulomatose avec polyangéite (GPA, anciennement granulomatose de Wegener) ;
- la micropolyangéite (MPA) ou polyangéite microscopique ;
- la granulomatose éosinophile avec polyangéite (EGPA) (anciennement syndrome de Churg et Strauss) ;
- la glomérulonéphrite pauci-immune isolée.
Anticorps antiphospholipides
La recherche des anticorps antiphospholipides doit être réalisée dans les situations évocatrices d’un syndrome des antiphospholipides (primaire ou associé) [- la sérologie syphilitique : TPHA-VDRL, avec un VDRL positif et un TPHA négatif (fausse sérologie syphilitique positive) ;
- un test fonctionnel, à la recherche d’un anticoagulant circulant lupique (appelé aussi antiprothrombinase) : temps de céphaline activée (TCA) allongé et absence de correction de cet allongement par l’adjonction de plasma témoin ;
- anticorps anticardiolipines (IgG et/ou IgM) présents à au moins 2 reprises, à un titre intermédiaire ou élevé (> 40 UGPL ou MPL, ou > 99e percentile) [ELISA] ;
- anticorps anti-bêta 2 GP1 (IgG et/ou IgM) présents à un titre > au 99e percentile, à au moins 2 reprises, à 12 semaines d’intervalle (ELISA).
Hypergammaglobulinémie polyclonale
La constatation d’une hypergammaglobulinémie polyclonale nécessite la recherche de son origine et n’est pas spécifique des pathologies auto-immunes. Les causes sont :
- les infections chroniques : bactériennes (endocardites, abcès profonds, tuberculose), virales (VIH, VHB, VHC, CMV, EBV), mycoses, paludisme et leishmaniose ;
- les hépatopathies chroniques (alcooliques, virales, auto-immunes, granulomateuses) : bloc β γ ;
- les lymphomes ;
- les maladies auto-immunes systémiques.
Cryoglobulinémie
- cryoglobulines monoclonales (de type I), le plus souvent IgM ; sont associées à une hémopathie B (leucémie lymphoïde chronique, lymphome, maladie de Waldenström) ; un myélome ou une gammapathie de signification indéterminée (MGUS) ;
- cryoglobulines mixtes (de type II et III), car composées d’immunoglobulines polyclonales associées à une immunoglobuline monoclonale (type 2) ou non (type 3). Elles peuvent être à l’origine de vascularites à complexes immuns (vascularites cryoglobulinémiques) parfois sévères, avec atteinte multiviscérale comprenant le plus souvent une atteinte cutanée, neurologique périphérique et rénale. S’y associe un effondrement de la fraction C4 du complément sérique et du CH50 avec une fraction C3 le plus souvent normale. Les cryoglobulinémies mixtes sont majoritairement liées à l’infection chronique par le virus de l’hépatite C, plus rarement aux connectivites (syndrome de Gougerot-Sjögren, lupus érythémateux systémique) et aux lymphomes B.
Principes de traitement et surveillance au long terme d’une maladie auto-immune
- à court terme : contrôler la symptomatologie, préserver le pronostic fonctionnel lié à l’atteinte d’un organe, préserver les fonctions vitales dans les formes graves ;
- à moyen terme : s’opposer à l’évolution prévisible des atteintes viscérales, prévenir les poussées, préserver l’insertion professionnelle du patient et la qualité de vie ;
- à long terme : limiter les séquelles et les effets secondaires des traitements.
- l’éducation du patient concernant la maladie, les traitements, les mesures hygiéno-diététiques éventuelles, les risques potentiels liés à la grossesse, le calendrier vaccinal, etc. ;
- les traitements médicamenteux : le traitement médical peut être distingué en traitement symptomatique (AINS, colchicine) et traitement immunomodulateur ou immunosuppresseur.
Le traitement de fond permettant le contrôle de la maladie auto- immune fait appel habituellement à l’immunosuppression et/ou à l’immunomodulation. Certains médicaments comme l’hydroxychloroquine ou les immunoglobulines intraveineuses ont un mécanisme de contrôle de la réponse immunitaire qui ne diminue pas cette réponse.
Parmi les immunosuppresseurs, il faut distinguer les immunosuppresseurs conventionnels et les biothérapies, ayant une action plus ciblée contre un composant du système immunitaire. Les modalités d’utilisation des immunosuppresseurs (indications, doses, durée, association de plusieurs molécules) dépendent de la maladie concernée, le type et le nombre d’atteintes et le risque vital éventuel. Le traitement comprend habituellement un traitement d’attaque lors de la phase aiguë, puis le traitement d’entretien de durée variable et souvent mal codifié.
Les différentes classes médicamenteuses sont (
- les corticoïdes (per os ou en bolus intraveineux) ;
- les antipaludéens de synthèse (hydroxychloroquine, chloroquine) ;
- les immunosuppresseurs classiques, en particulier les antimitotiques (méthotrexate, azathioprine, cyclophosphamide, ciclosporine, mycophénolate mofétil) ;
- les immunoglobulines intraveineuses ;
- les biothérapies ciblées : antagonistes du TNFα (infliximab, étanercept, adalimumab, certolizumab, golimumab), anti-IL1 (anakinra, canakinumab), antagonistes IL6-R (tocilizumab), anticorps anti-CD20 (rituximab) [v. Item 198].• ›››
La présence d’un autoanticorps doit être interprétée en fonction du contexte clinique, du type d’autoanticorps et de sa spécificité, de potentiels médicaments inducteurs ; la présence d’un autoanticorps n’est pas synonyme de maladie auto-immune, et les autoanticorps peuvent être absents dans certains cas de maladie auto-immune.
Les principes de traitement d’une maladie auto-immune doivent comprendre les mesures médicamenteuses, l’éducation et l’information et un objectif à court terme (contrôle de la maladie), à moyen terme (éviter les poussées et favoriser l’insertion du patient et la qualité de vie) et à long terme (éviter les séquelles et les effets secondaires).
A. Mekinian déclare avoir été pris en charge lors de congrès par LFB, Genzyme et Chugai. O. Fain déclare être conseiller scientifique auprès des laboratoires Shire et Behring et avoir reçu des financements pour des congrès à l’étranger de ces mêmes laboratoires ainsi que de GSK, Pfizer et Novartis.
FIGURE
Les pathologies auto-immunes sont séparées en maladie d’organes, avec l’atteinte d’un organe et la présence d’un autoanticorps spécifique, et des maladies systémiques, avec des anticorps qui ne sont pas spécifique d’organe et une atteinte qui peut toucher plusieurs organes. La présence d’un autoanticorps doit être interprétée en fonction du contexte clinique, du type d’autoanticorps et de sa spécificité, de potentiels médicaments inducteurs ; la présence d’un autoanticorps n’est pas synonyme de maladie auto-immune, et les autoanticorps peuvent être absents dans certains cas de maladie auto-immune. Les principes de traitement d’une maladie auto-immune doivent comprendre les mesures médicamenteuses, l’éducation et l’information et un objectif à court terme (contrôle de la maladie), à moyen terme (éviter les poussées et favoriser l’insertion du patient et la qualité de vie) et à long terme (éviter les séquelles et les effets secondaires).
Dans cet article
 Encadrés
Encadrés
Tableau 2
|
Maladies auto-immunes non spécifiques d’organes
|
|
Syndrome Gougerot-Sjögren Myosites inflammatoires Sclérodermie Lupus érythémateux systémique Vascularites systémiques
|
Tableau 4
| |
Circonstances justifiant la recherche des anticorps anticytoplasme des polynucléaires
| |
|
Purpura vasculaire (infiltré) Nécrose cutanée Gangrène ou ulcère digital Asthme sévère et/ou tardif Myalgies
|
Arthralgies inexpliquées d’horaire inflammatoire Mono- ou polyneuropathie Syndrome pneumorénal Hématurie et/ou protéinurie Syndrome inflammatoire inexpliqué
|
Tableau 5
| |
Circonstances justifiant la recherche des anticorps antiphospholipides
| |
|
Thromboses de siège inhabituel (veine cave, sus-hépatique, autre) Thrombose artérielle si âge < 45 ans Thrombose veineuse si âge < 45 ans Avortements spontanés précoces ≥ 3
|
Mort fœtale (> 10 SA [semaines d'aménorrhée]) Prééclampsie ou éclampsie < 34 SA Livedo racemosa Thrombopénie inexpliquée TPHA négatif-VDRL positif Lupus érythémateux systémique
|