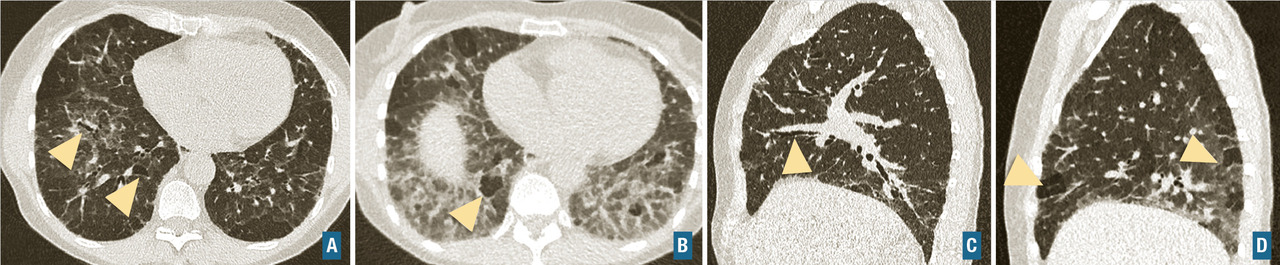
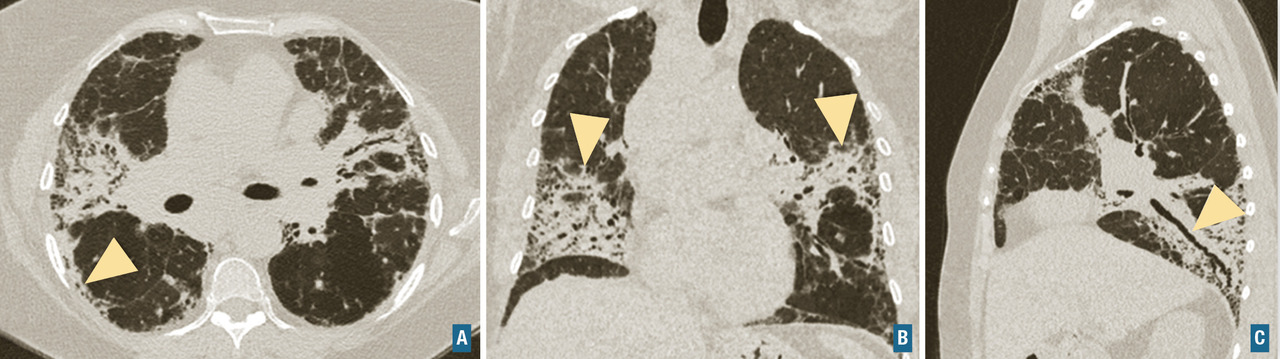
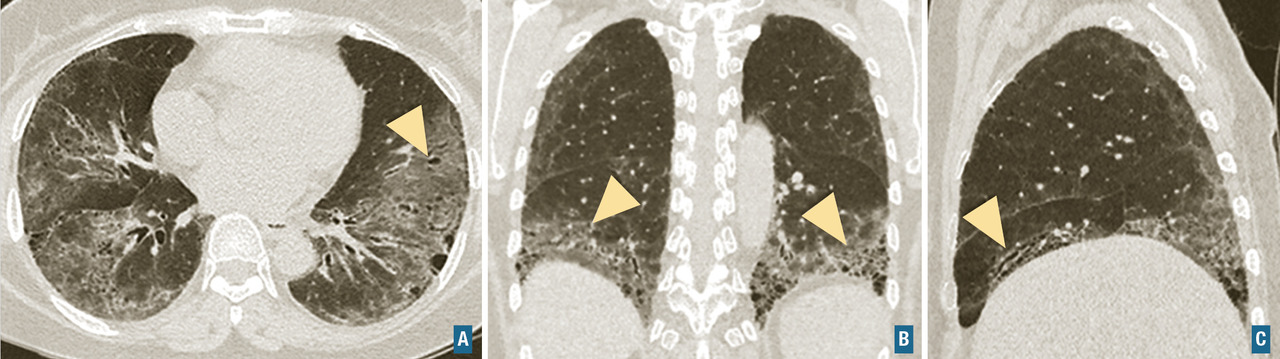
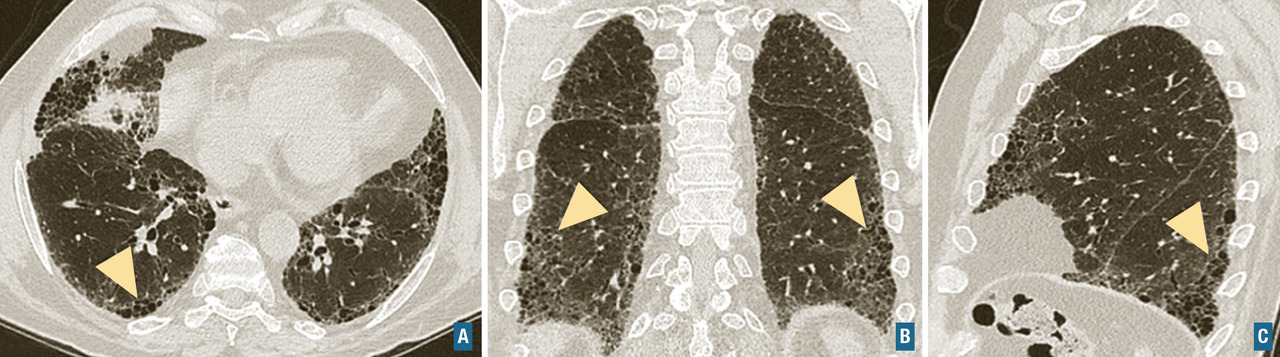
Diagnostiquer une pneumopathie interstitielle diffuse.
Définition
Épidémiologie
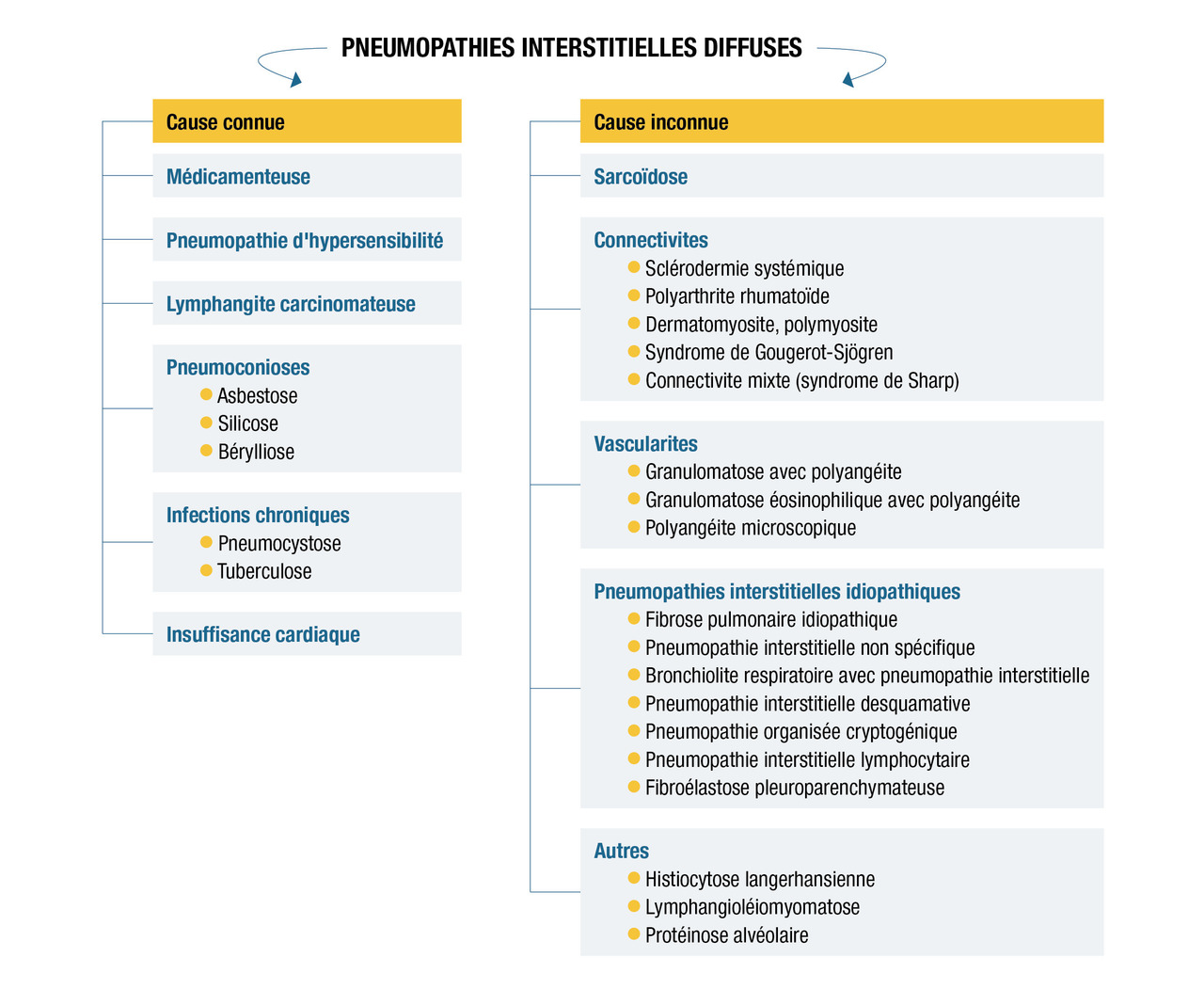
Classification
Les causes hémodynamiques et infectieuses dominent les pneumopathies interstitielles aiguës tandis que les causes des pneumopathies interstitielles diffuses subaiguës/chroniques sont souvent plus variées et de diagnostic plus difficile.
Les pneumopathies interstitielles diffuses subaiguës/chroniques peuvent être divisées selon que le contexte étiologique est connu (exposition environnementale ou professionnelle, prise médicamenteuse, néoplasie connue, etc.) ou inconnu.
Pneumopathies interstitielles diffuses subaiguës et chroniques
Démarche diagnostique
Interrogatoire
L’interrogatoire s’attache à relever les antécédents pertinents : insuffisance cardiaque, connectivite, cancer évolutif, immunodépression, toxicomanie, etc. On recherche une prise médicamenteuse, une exposition professionnelle ou de loisir à des aérocontaminants : amiante, silice, poussières organiques ou de métaux, etc.Examen clinique
Il convient de préciser les symptômes et signes fonctionnels (- respiratoires : dyspnée et mode d’installation, toux, expectorations, douleur thoracique, hémoptysie, sifflements intrathoraciques, crépitants et notamment crépitants secs type « Velcro » ;
- cardiovasculaires : signes d’insuffisance cardiaque (orthopnée, œdèmes des membres inférieurs, turgescence jugulaire et reflux hépatojugulaire) ;
- signes évocateurs de maladie de système ou granulomatose : syndrome sec, uvéite, arthralgies inflammatoires, éruption cutanée (mains de mécanicien, érythème liliacé des paupières, érythème noueux…), phénomène de Raynaud, sclérodactylie, myalgies et faiblesse musculaire, mononeuropathie unique ou multiple, rhinite croûteuse, etc.
Examens biologiques
Le bilan biologique doit comporter une numération formule sanguine et un bilan immunologique comprenant au minimum des anticorps antinucléaires et souvent des anticorps anticytoplasme des polynucléaires neutrophiles (ANCA) ainsi que leur spécificité en cas de positivité. On recherche également une hyperéosinophilie, une hypercalcémie, une hypergammaglobulinémie ou un pic monoclonal, une insuffisance rénale.Des explorations plus spécifiques sont demandées selon l’orientation étiologique : auto-anticorps des myosites (dont les antisynthétases), anticorps antipeptide cyclique citrulliné, facteur rhumatoïde, calciurie, précipitines sériques, etc.
Examens radiologiques
Radiographie pulmonaire
Scanner thoracique
Explorations fonctionnelles respiratoires
Elles objectivent le plus souvent un syndrome restrictif (baisse de la capacité vitale et de la capacité pulmonaire totale), notamment dans les pneumopathies interstitielles diffuses fibrosantes. Un trouble ventilatoire obstructif peut s’associer, ou prédominer, dans certaines pathologies : pneumopathie d’hypersensibilité, histiocytose langerhansienne, lymphangioléiomyomatose, ou sarcoïdose. Le coefficient de transfert pulmonaire du monoxyde de carbone (KCO) est diminué, traduisant l’altération de la diffusion alvéolocapillaire en oxygène. La pression artérielle en oxygène (PaO2) peut être normale, diminuée à l’effort (mise en évidence par une désaturation au test de marche ou une épreuve d’effort) ou au repos (hypoxémie de repos).Endoscopie bronchique
Elle permet la réalisation du lavage broncho-alvéolaire, de biopsies bronchiques ou transbronchiques à la pince, de biopsies d’adénopathies médiastinales échoguidées (recherche de sarcoïdose, de néoplasie), et dans certains centres de cryobiopsies transbronchiques. Le lavage broncho-alvéolaire est de préférence réalisé dans un territoire orienté par l’imagerie. L’aspect macroscopique peut orienter le diagnostic : rosé ou rouge (hémorragie intra-alvéolaire), ou laiteux (protéinose alvéolaire). La recherche d’agents infectieux doit être systématique. La formule cytologique (Place de l’anatomopathologie
Lors de l’endoscopie bronchique :- les biopsies bronchiques (étagées, systématiques ou dirigées par l’imagerie ou par un aspect macroscopique anormal) peuvent confirmer un diagnostic de sarcoïdose ou de lymphangite carcinomateuse ;
- les biopsies transbronchiques à la pince prélèvent un petit échantillon du parenchyme pulmonaire et sont utiles dans la sarcoïdose, la pneumopathie organisée cryptogénique ou la lymphangite carcinomateuse. Elles peuvent parfois occasionner un pneumothorax ou une hémoptysie de faible abondance ;
- les cryobiopsies transbronchiques utilisent une sonde cryogénique pour geler le parenchyme pulmonaire au contact et obtenir un prélèvement histologique de 5 à 15 mm de grand axe. Elles offriraient une rentabilité diagnostique correcte pour une morbi-mortalité inférieure à celle de la biopsie pulmonaire chirurgicale. Leur faible disponibilité (quelques centres en France) fait que leur place dans la stratégie diagnostique des pneumopathies interstitielles diffuses reste encore à définir ;
- la ponction échoguidée transbronchique ou transœsophagienne d’une adénopathie médiastinale ou hilaire permet le diagnostic de sarcoïdose, tuberculose ganglionnaire, néoplasie ou lymphome.
- une médiastinoscopie sus-sternale constitue un moyen peu invasif d’obtenir un prélèvement d’adénopathies paratrachéales droites ou médiastinales ;
- la biopsie pulmonaire chirurgicale par vidéothoracoscopie permet d’obtenir des prélèvements provenant de lobes différents. Elle n’est pas dénuée de risque, avec une mortalité d’environ 1 à 2 % et une morbidité proche de 10 %. Chaque indication doit faire l’objet d’une discussion multidisciplinaire en milieu spécialisé. Elle n’est envisagée que si la fonction respiratoire du patient le permet, etuniquement si des conséquences sur la prise en charge sont escomptées. Cette technique, actuellement moins utilisée pour le diagnostic de la fibrose pulmonaire idiopathique grâce à l’amélioration des connaissances, reste utile dans les cas difficiles en l’absence de facteurs de risque.
- biopsie cutanée d’une suspicion de sarcoïde (sarcoïdose) ;
- biopsie des glandes salivaires accessoires (sarcoïdose, syndrome de Gougerot-Sjögren) ;
- biopsie d’autres organes (métastases d’une néoplasie pulmonaire) ;
- biopsie musculaire (myosites), etc.
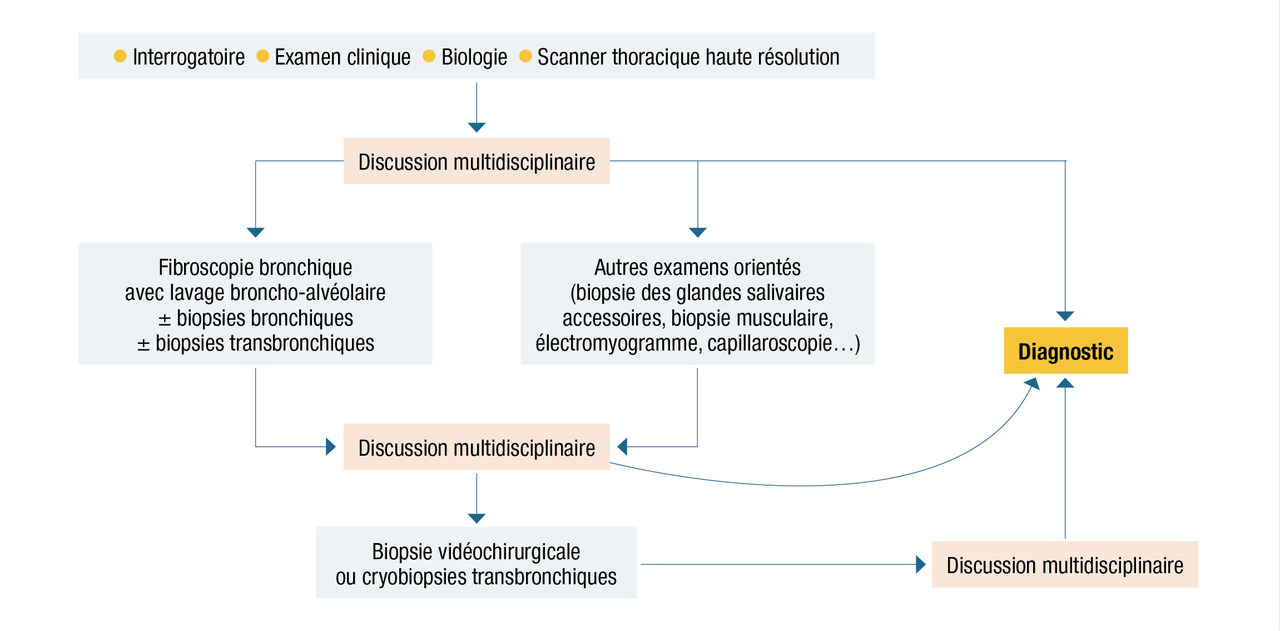
Synthèse diagnostique
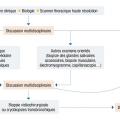
Le bilan de première intention comprend un interrogatoire et un examen clinique rigoureux, des explorations fonctionnelles respiratoires, un scanner thoracique haute résolution, un bilan biologique, et souvent une endoscopie bronchique avec lavage broncho-alvéolaire. Les prélèvements histologiques sont réalisés au cas par cas. Au terme du bilan de première intention, il est recommandé d’effectuer la synthèse de chaque cas en discussion multidisciplinaire, comprenant des cliniciens, des radiologues et des anatomopathologistes expérimentés dans le domaine des pneumopathies interstitielles diffuses. La démarche diagnostique est résumée dans laÉtiologie
Pneumopathies interstitielles diffuses de cause connue
Médicamenteuses
Il convient de retenir qu’une cause médicamenteuse doit toujours être évoquée lors du diagnostic d’une pneumopathie interstitielle diffuse, en s’aidant au besoin d’une base de données en ligne (www.pneumotox.com).
Pneumopathie d’hypersensibilité
L’interrogatoire doit être précis lors de la recherche d’une exposition : oiseaux, foin surtout s’il est moisi, moisissures domestiques (jacuzzi, humidité, dégâts des eaux), activité professionnelle. Un questionnaire standardisé (par exemple, liste du GERM « O » P des causes de pneumopathie d’hypersensibilité) peut être utile pour préciser une exposition. La recherche de précipitines sériques dirigées contre des antigènes suspectés peut confirmer une exposition, mais n’est ni nécessaire ni suffisante au diagnostic.
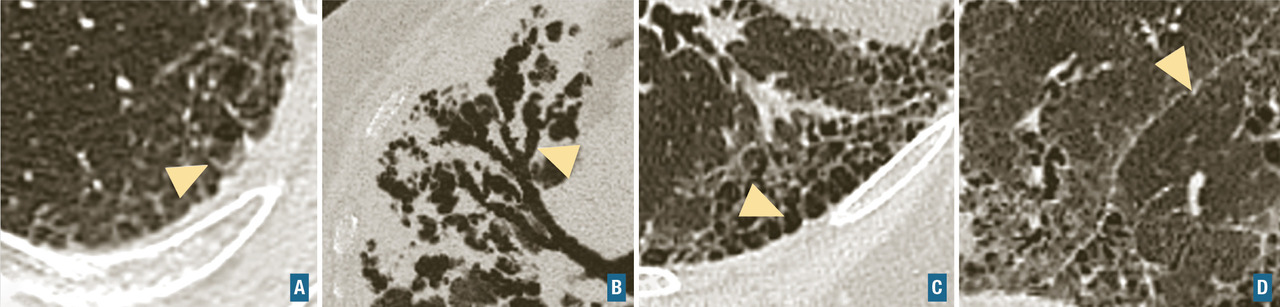
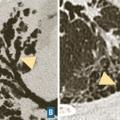
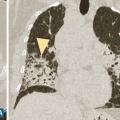
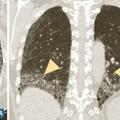
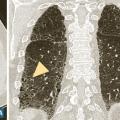
Les symptômes comprennent une dyspnée, une toux, des râles « piaulants » (squeaks) à l’auscultation et parfois des signes généraux semi-retardés : frissons, asthénie, fièvre. Le scanner thoracique met en évidence une atténuation en mosaïque à trois niveaux de densité, des plages de verre dépoli et des nodules flous centrés sur les bronches (
Le traitement repose sur l’éviction de l’agent causal et sur la corticothérapie.
Pneumoconioses
L’asbestose est la plus fréquente des pneumoconioses et est secondaire à une exposition importante à l’amiante. Les professions les plus touchées sont les travailleurs sur chantier naval, les employés du bâtiment, les plombiers et les chauffagistes. L’atteinte pulmonaire ressemble à celle de la fibrose pulmonaire idiopathique, la distinction entre les deux est facilitée en cas de présence de plaques pleurales, calcifiées ou non, marqueurs de l’exposition asbestosique. Des corps asbestosiques peuvent être trouvés dans l’expectoration, le lavage broncho-alvéolaire ou les prélèvements histologiques. L’asbestose peut faire l’objet d’une indemnisation par le Fonds d’indemnisation des victimes de l’amiante (FIVA). L’exposition à l’amiante augmente également le risque de cancer bronchique et de mésothéliome malin.
Silicose : pneumoconiose historique des mineurs de fond, elle concerne les professions avec activité de taille, sablage ou ponçage de pierre : tunneliers, fonderie-métallurgie, carrière de pierre, industrie du verre, bâtiment, prothésistes dentaires, fabrication de brique et céramique… L’accumulation de particules de silice cristalline dans les voies aériennes pendant une longue période (> 10 ans) aboutit à une atteinte pulmonaire nodulaire et fibrosante. L’aspect radiologique est celui d’une pneumopathie interstitielle diffuse avec opacités nodulaires bilatérales et symétriques des sommets, de taille variable : micronodules bien définis (3-5 mm), nodules (aspect de « tempête de neige »), masses asymétriques et confluentes responsables d’une atteinte fibrosante avec distorsion architecturale. Des adénopathies médiastinales peuvent être présentes, typiquement calcifiées « en coquille d’œuf ». Cet aspect peut faire discuter une sarcoïdose, mais le diagnostic se passe habituellement de biopsie en présence d’une atteinte pulmonaire évocatrice associée à une exposition importante et prolongée. La silicose peut s’associer à une connectivite, réalisant le syndrome d’Erasmus (sclérodermie systémique) ou de Caplan-Colinet (polyarthrite rhumatoïde).
La bérylliose chronique est caractérisée par une atteinte pulmonaire granulomateuse mimant une sarcoïdose. Elle touche les patients présentant une susceptibilité génétique (phénotype HLA) et une exposition au béryllium : prothésistes dentaires, mécaniciens, industrie aérospatiale, horlogerie. Le scanner thoracique montre une pneumopathie interstitielle diffuse micronodulaire de distribution lymphatique avec opacités en verre dépoli et adénopathies hilaires. Le diagnostic repose sur la mise en évidence d’une exposition et un test de transformation lymphoblastique positif en présence de béryllium.
Pneumoconioses de surcharge : elles rassemblent les affections pulmonaires secondaires à l’inhalation prolongée d’autres particules inertes (fer, talc, titane, baryum…). L’atteinte radiologique est variable car dépendante de la densité des particules.
Lymphangite carcinomateuse
Autres
Pneumopathies interstitielles diffuses de cause inconnue
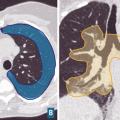
Sarcoïdose
L’aspect scannographique rencontré au cours de la sarcoïdose est décrit dans la
Connectivites
Vascularites
Syndrome de Goodpasture : c'est une vascularite des petits vaisseaux avec anticorps antimembrane basale glomérulaire (MBG). C’est une cause de syndrome « pneumorénal » qui associe une hémorragie intra-alvéolaire et une néphropathie glomérulaire.
Pneumopathies interstitielles diffuses idiopathiques
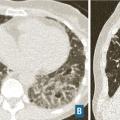
Pneumopathie interstitielle non spécifique (PINS) idiopathique : c’est une maladie rare qui constitue la forme idiopathique du tableau de PINS observé dans plusieurs autres maladies (connectivites, pneumopathies médicamenteuses, fibroses pulmonaires familiales). L’aspect de PINS au scanner est celui d’une pneumopathie interstitielle diffuse associant de manière variable des réticulations, du verre dépoli et des signes de fibrose (bronchectasies et bronchiolectasies par traction), le plus souvent sans rayon de miel. L’épargne de la corticalité pulmonaire sur environ 5-10 mm et une prédominance centrale sont évocatrices. Histologiquement, on note un infiltrat interstitiel inflammatoire et des lésions de fibrose, en proportions variables.
Pneumopathies interstitielles diffuses idiopathiques liées au tabac : la bronchiolite respiratoire avec pneumopathie interstitielle diffuse et la pneumopathie interstitielle desquamative sont liées à l’accumulation de macrophages contenant des pigments tabagiques bruns.
Pneumopathie interstitielle lymphocytaire : c’est une entité rare, le plus souvent secondaire à un syndrome de Gougerot-Sjögren mais parfois à une maladie auto-immune ou un état dysimmunitaire (déficit immunitaire combiné sévère, sida).
Pneumopathies interstitielles diffuses inclassables : dans 15 % des cas, et malgré la discussion multidisciplinaire, il n’est pas possible d’aboutir à un diagnostic. Cela peut être dû à des données incomplètes (par exemple l’impossibilité de réaliser une biopsie pulmonaire), discordantes, ou à un traitement préalable qui modifierait l’aspect radiologique ou histologique. Ces pneumopathies interstitielles diffuses sont dites « inclassables », et leur prise en charge doit se faire selon le diagnostic provisoire retenu (« diagnostic de travail ») et l’évolution clinique.
Autres
Protéinose alvéolaire, lymphangioléiomyomatose, fibroélastose pleuroparenchymateuse, etc.
Discussion multidisciplinaire
L’efficacité de la discussion multidisciplinaire a été attestée dans des études sur les performances diagnostiques et la reproductibilité inter-observateurs, et constitue aujourd’hui un élément incontournable de la démarche diagnostique et thérapeutique des pneumopathies interstitielles diffuses.
Pneumopathies interstitielles diffuses aiguës
Contexte du diagnostic
Étiologie
Œdème pulmonaire cardiogénique
L’insuffisance ventriculaire gauche peut compliquer l’évolution de toutes les cardiopathies gauches et peut être responsable d’une pneumopathie interstitielle diffuse en cas d’œdème interstitiel.Œdème pulmonaire lésionnel
Il se caractérise histologiquement par un aspect de dommage alvéolaire diffus et traduit une agression aiguë du poumon par un agent inhalé (toxique, liquide gastrique), une infection, un médicament, un agent toxique par voie systémique, un état inflammatoire aigu systémique (pancréatite aiguë, coagulation intravasculaire disséminée, état de choc septique, hémorragique, anaphylactique…).Infections pulmonaires
Le diagnostic est porté par la documentation microbiologique sur le lavage broncho-alvéolaire ou les autres prélèvements respiratoires (aspiration bronchique, examens cytobactériologique et mycologique des expectorations, prélèvement distal protégé chez le patient intubé). Les agents incriminés sont bactériens (agents « atypiques » comme Chlamydophila pneumoniae et Mycoplasma pneumoniae, légionellose, infection à Coxiella burnetii, miliaire tuberculeuse), viraux (grippe, coronavirus, virus respiratoire syncytial) ou fongiques (principalement Pneumocystis jiroveci).Pneumopathies médicamenteuses
Les molécules les plus fréquemment incriminées sont les bêtalactamines, l’amiodarone, le méthotrexate et la nitrofurantoïne. Il s’agit le plus souvent d’atteinte mimant une pneumopathie infectieuse. Les lésions au scanner sont variables, et l’interrogatoire doit préciser la chronologie d’introduction des médicaments. La prise en charge consiste à retirer le ou les médicaments suspects et nécessite parfois l’administration de corticoïdes.Pneumopathies d’hypersensibilité aiguës
Les pneumopathies d’hypersensibilité aiguës peuvent se présenter sur un mode aigu ou suraigu, principalement lors de l’inhalation d’une quantité massive d’antigène.Hémorragie intra-alvéolaire
Elle est définie par un liquide hémorragique au lavage broncho-alvéolaire dont la coloration s’accentue au fil des aliquots ou après 48 heures par un score de Golde > 100 (fondé sur le compte des sidérophages à la coloration de Perls). La présentation clinique est variable selon l’étiologie, une hémoptysie peut être présente. Le scanner thoracique met en évidence des plages de condensation alvéolaire ou de verre dépoli selon l’intensité. Les causes sont variées : vascularites (syndrome de Goodpasture, GPA, PAM), connectivites, infections (leptospirose), toxicité médicamenteuse (nitrofurantoïne, pénicillamines, abciximab, bévacizumab, acide rétinoïque, erlotinib, thrombolytiques), insuffisance cardiaque gauche, thrombopénie ou coagulopathie. La prise en charge est conditionnée par l’étiologie.Exacerbation aiguë d’une pneumopathie interstitielle diffuse chronique
Les pneumopathies interstitielles diffuses fibrosantes et notamment la fibrose pulmonaire idiopathique peuvent rarement être découvertes à l’occasion d’une poussée de la maladie. Le diagnostic d’exacerbation de fibrose est posé sur une majoration récente des symptômes respiratoires et l’apparition de nouvelles opacités en verre dépoli au scanner thoracique, sur un aspect sous-jacent de maladie fibrosante. La prise en charge consiste à identifier et traiter un facteur déclenchant ou surajouté (embolie pulmonaire, infection, inhalation, cause médicamenteuse ou biopsie pulmonaire) et en l’administration de corticoïdes intraveineux à haute dose suivie d’un relais per os et d’une décroissance progressive.Autres causes
Pneumopathie interstitielle aiguë
La pneumopathie interstitielle aiguë (anciennement syndrome de Hamman-Rich) est une forme idiopathique de syndrome de détresse respiratoire aiguë (SDRA). La prise en charge nécessite fréquemment le recours à la ventilation. Il est capital d’éliminer une cause, notamment infectieuse ou métabolique. La biopsie pulmonaire rarement réalisée retrouve un aspect de dommage alvéolaire diffus.Pneumopathie organisée cryptogénique
Le terme de pneumopathie organisée décrit un aspect histologique (infiltrat inflammatoire et bourgeon fibroconjonctif dans les alvéoles) d’origine majoritairement secondaire (infection, médicaments, connectivites). La pneumopathie organisée cryptogénique est une entité mimant une pneumopathie infectieuse (dyspnée, toux, crépitants, signes généraux) d’installation aiguë ou subaiguë, avec au scanner thoracique des plages de condensation alvéolaire, bilatérales, souvent migratrices, auxquelles s’associent fréquemment des opacités en verre dépoli. Le diagnostic peut être confirmé par des biopsies transbronchiques ou une biopsie transpariétale. L’évolution est favorable sous corticothérapie. Les rechutes sont fréquentes.Conclusion
POINTS FORTS À RETENIR
Les pneumopathies interstitielles diffuses regroupent de très nombreuses entités différentes, soulignant l’importance d’une démarche diagnostique rationnelle. La définition est radiologique, avec des opacités pulmonaires bilatérales.
Les pneumopathies interstitielles diffuses aiguës sont le plus souvent causées par des infections ou une insuffisance cardiaque, et ces causes doivent être recherchées en priorité. La prise en charge se fait parfois en réanimation.
L’interrogatoire, l’examen clinique et la recherche d’expositions médicamenteuses, toxiques, professionnelles ou domestiques sont des éléments cruciaux pour aboutir à un diagnostic.
La sarcoïdose, la fibrose pulmonaire idiopathique, les connectivites, et les pneumopathies d’hypersensibilité dominent les causes des pneumopathies interstitielles diffuses subaiguës/chroniques. La démarche diagnostique doit être rigoureuse et faire appel à une discussion multidisciplinaire, si possible dans un centre de compétence.
Le recours à la biopsie pulmonaire chirurgicale est plus rare qu’auparavant mais conserve des indications.
Pneumopathie interstitielle diffuse
L’item 206/210 apparaît souvent aux étudiants comme un chapitre « catalogue », avec un grand nombre de pathologies dont il est parfois difficile de faire la distinction car elles ne sauraient être toutes décrites précisément en un seul chapitre.
Cet item pourrait cependant faire l’objet d’un dossier ou de questions isolées au concours, notamment sur les situations les plus fréquentes :
– sarcoïdose (item spécifique de l’ECN) ;
– fibrose pulmonaire idiopathique ;
– lymphangite carcinomateuse ;
– insuffisance cardiaque ;
– connectivites.
La sarcoïdose ou la fibrose pulmonaire idiopathique peuvent constituer un dossier entier. La lymphangite carcinomateuse conclura un dossier de cancérologie (par exemple l’interprétation d’un scanner thoracique) ; l’insuffisance cardiaque sera comprise dans un dossier de cardiologie. Il peut être attendu d’évoquer une pneumopathie médicamenteuse ou d’hypersensibilité.
Dans un dossier de connectivite, l’apparition d’une pneumopathie interstitielle diffuse devra faire discuter :
– une complication des traitements (méthotrexate) ;
– une infection opportuniste ;
– une atteinte pulmonaire de la maladie causale.
Dans un dossier de diagnostic de pneumopathie interstitielle diffuse, il conviendra de mémoriser les éléments clés :
– l’importance de l’âge et du sexe (typiquement homme de plus de 60 ans pour la fibrose pulmonaire idiopathique, femme de moins de 50 ans pour une connectivite) ;
– l’importance de l’interrogatoire, notamment sur les expositions (médicamenteuses, professionnelles ou domestiques) ;
– les signes cliniques évocateurs de pathologie sous-jacente (syndrome sec, phénomène de Raynaud, etc.) ;
– l’interprétation du scanner thoracique, qui sera typique dans un dossier de fibrose pulmonaire idiopathique par exemple ;
– discuter le diagnostic différentiel.
Une question au concours sur ce chapitre se prête également bien à l’évaluation des connaissances en pneumologie de l’étudiant : interprétation de gaz du sang, sémiologie clinique et radiologique, lavage broncho-alvéolaire, etc.
Cottin V, Crestani B, Cadranel J, et al. French practical guidelines for the diagnosis and management of idiopathic pulmonary fibrosis.2017 update. Short-length version. Rev Mal Respir 2017 Sep 21. pii: S0761-8425(17)30209-7. doi: 10.1016/j.rmr.2017.07.018. French.
Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung Diseases. N Engl J Med 2020;383(10):958-68. doi: 10.1056/NEJMra2005230. PMID: 32877584.
 Encadrés
Encadrés