Les premiers signes de la polychondrite chronique atrophiante, aussi bien cliniques que biologiques, sont inconstants. Cette pathologie constitue donc un véritable défi diagnostique. Il existe trois principaux phénotypes associés à des pronostics différents imposant une prise en charge spécifique. La réalisation d’examens d’imagerie permet d’évaluer les atteintes d’organes et oriente vers le traitement le plus adapté.
La polychondrite chronique atrophiante (PCA) est une maladie rare caractérisée par la survenue d’une inflammation des cartilages, appelée chondrite. Il s’agit d’une maladie évoluant par poussées touchant en particulier la sphère ORL (pavillon des oreilles, cartilages du nez) mais également l’arbre trachéobronchi–que. Elle constitue une véritable maladie systémique car elle peut également concerner d’autres organes tels que l’œil, le système cardiovasculaire ou encore la peau. La PCA est associée dans 30 % des cas à une autre maladie auto-immune ou inflammatoire, qu’il convient de dépister. Par ailleurs, une forme particulière a été décrite récemment : il est important de savoir la reconnaître compte tenu de son association à des hémopathies (essentiellement syndrome myélodysplasique) chez les hommes de plus de 50 ans.
Le diagnostic de PCA doit être évoqué devant la survenue de chondrites, qui peuvent être absentes au début de la maladie et n’apparaître qu’au bout de plusieurs années d’évolution. La PCA débute habituellement autour de 40 à 45 ans, mais peut survenir à tout âge puisqu’il existe d’exceptionnelles formes pédiatriques. Le diagnostic de cette maladie rare est d’autant plus difficile qu’il n’existe aucun marqueur biologique spécifique, et que le syndrome inflammatoire biologique peut être absent lors des poussées dans près de 40 % des cas. Elle constitue donc un véritable défi diagnostique, nécessitant notamment d’éliminer des pathologies comme les vascularites à ANCA (anticorps anticytoplasme des polynucléaires neutrophiles). Certains examens d’imagerie spécifiques s’imposent pour évaluer au mieux les atteintes d’organes associées et discuter du traitement le plus adapté.
Le diagnostic de PCA doit être évoqué devant la survenue de chondrites, qui peuvent être absentes au début de la maladie et n’apparaître qu’au bout de plusieurs années d’évolution. La PCA débute habituellement autour de 40 à 45 ans, mais peut survenir à tout âge puisqu’il existe d’exceptionnelles formes pédiatriques. Le diagnostic de cette maladie rare est d’autant plus difficile qu’il n’existe aucun marqueur biologique spécifique, et que le syndrome inflammatoire biologique peut être absent lors des poussées dans près de 40 % des cas. Elle constitue donc un véritable défi diagnostique, nécessitant notamment d’éliminer des pathologies comme les vascularites à ANCA (anticorps anticytoplasme des polynucléaires neutrophiles). Certains examens d’imagerie spécifiques s’imposent pour évaluer au mieux les atteintes d’organes associées et discuter du traitement le plus adapté.
Maladie rare, mal répertoriée
L’épidémiologie de la PCA est mal connue, et il n’existe à ce jour aucun registre national ou international de la maladie.1,2 Rare, mais probablement sous-diagnostiquée, son incidence est de l’ordre de 1 à 3 cas par million par an.3-5 La maladie débute en général chez des sujets d’âge moyen, aux alentours de 40 ans dans les séries récentes,4,6,7 même si de très rares cas pédiatriques sont rapportés.8,9 Le sex-ratio est proche de 1 dans la plupart des séries.6,7
D’après une étude japonaise récente,10 la mortalité de la maladie a diminué de 9,2 % en 2009 à 1,6 % en 2019, possiblement du fait de l’utilisation plus fréquente des biothérapies, en particulier dans les formes avec atteintes trachéales. Dans une série hongroise,4 la survie à cinq ans était proche de celle de la population générale compte tenu de l’âge de révélation de la maladie, cependant inférieure à celle rapportée dans une série française (environ 95 % à cinq ans et environ 90 % à dix ans).7 Dans une série britannique, le ratio de mortalité standardisé était de 2,16 (intervalle de confiance à 95 % [IC95 %] : 1,24-3,51) avec les atteintes respiratoires, cardiaques et les cancers rapportés comme principales causes de décès.
D’après une étude japonaise récente,10 la mortalité de la maladie a diminué de 9,2 % en 2009 à 1,6 % en 2019, possiblement du fait de l’utilisation plus fréquente des biothérapies, en particulier dans les formes avec atteintes trachéales. Dans une série hongroise,4 la survie à cinq ans était proche de celle de la population générale compte tenu de l’âge de révélation de la maladie, cependant inférieure à celle rapportée dans une série française (environ 95 % à cinq ans et environ 90 % à dix ans).7 Dans une série britannique, le ratio de mortalité standardisé était de 2,16 (intervalle de confiance à 95 % [IC95 %] : 1,24-3,51) avec les atteintes respiratoires, cardiaques et les cancers rapportés comme principales causes de décès.
À la frontière de l’immunité innée et adaptative
Comme beaucoup de maladies similaires, la PCA est associée génétiquement à certains allèles HLA,11 mais la recherche de ces allèles à risque ne présente pas d’intérêt en pratique courante. La chondrite semble être liée à une atteinte du tissu cartilagineux par un infiltrat inflammatoire avec des lymphocytes T autoréactifs dirigés contre certains antigènes spécifiques de cartilage12 et des monomacrophages, comme en témoignent des concentrations sériques élevées de monocytes chemoattractant protein 1 beta (MCP-1), de macrophages inflammatory protein 1-beta (MIP-1-beta) et d’interleukine 8 (IL-8).13 Différents auto-anticorps ont été mis en évidence, mais leur faible sensibilité fait que leur recherche n’est pas recommandée en pratique courante (notamment anticorps anticollagène de type 2 et matrilline-1).14,15 Il est intéressant de noter que l’immunisation contre la matrilline-1 (protéine spécifique du cartilage trachéal) reproduit l’atteinte respiratoire de la PCA chez le rat.16 Enfin, la forme de l’homme de plus de 50 ans associée aux hémopathies et notamment à un syndrome myélodysplasique (syndrome Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic [VEXAS]) présente une physiopathologie très particulière exposée infra.
Principaux phénotypes cliniques de PCA
L’analyse d’une série française de patients a permis de mettre en évidence trois principaux phénotypes cliniques :7 un phénotype de bon pronostic, avec principalement des chondrites mineures récurrentes ; une forme de moins bon pronostic caractérisée par l’atteinte de l’arbre trachéobronchique ; et un phénotype hématologique de pronostic encore sévère, qui a récemment été associé au syndrome VEXAS17,18 (tableau 1 ).
Présentation clinique typique : phénotype à chondrites mineures récurrentes
Dans 60 % des cas, la PCA se présente sous sa forme clinique évocatrice constituée par des chondrites du pavillon de l’oreille et du nez. Le diagnostic est relativement facile, après avoir éliminé les principaux diagnostics différentiels.
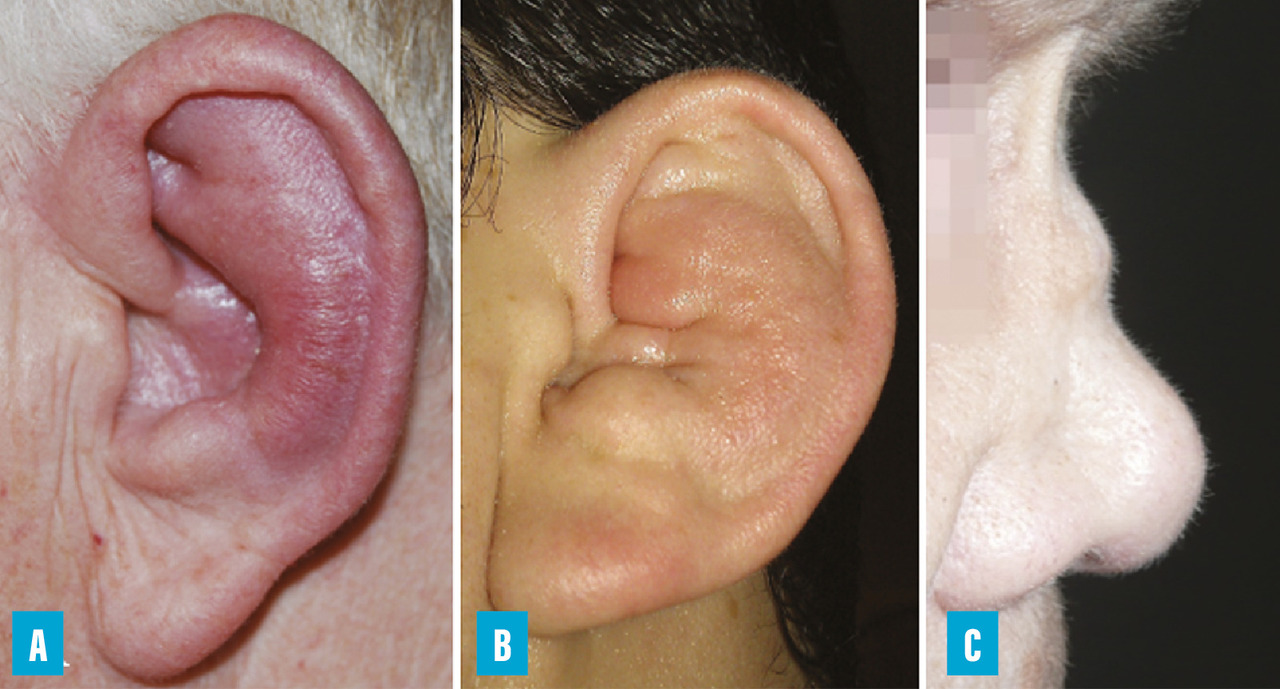
La chondrite du pavillon de l’oreille est la plus fréquente et la plus évocatrice de la maladie (jusqu’à 90 % des patients au cours de l’évolution) ; elle peut être uni- ou bilatérale et se traduit par une inflammation du cartilage du pavillon de l’oreille épargnant le lobule qui, lui, n’est pas cartilagineux (fig. 1A ) ; ce point est essentiel et permet de faire la différence avec d’autres pathologies de l’oreille, notamment infectieuses. Elle évolue durant quelques jours à quelques semaines, et peut régresser spontanément. En cas de poussées itératives, elle est responsable d’une destruction cartilagineuse en « oreille en chou-fleur » (fig. 1B ).
La chondrite nasale est moins fréquente (60 % des patients au cours de l’évolution de la maladie), caractérisée par une douleur au niveau de la zone de jonction entre l’os nasal et le cartilage au niveau de la racine du nez, généralement sans signes inflammatoires locaux. La répétition des poussées est également responsable d’une destruction du cartilage, avec une déformation typique mais non spécifique « en pied de marmite » (fig. 1C ).
La chondrite du pavillon de l’oreille est la plus fréquente et la plus évocatrice de la maladie (jusqu’à 90 % des patients au cours de l’évolution) ; elle peut être uni- ou bilatérale et se traduit par une inflammation du cartilage du pavillon de l’oreille épargnant le lobule qui, lui, n’est pas cartilagineux (
La chondrite nasale est moins fréquente (60 % des patients au cours de l’évolution de la maladie), caractérisée par une douleur au niveau de la zone de jonction entre l’os nasal et le cartilage au niveau de la racine du nez, généralement sans signes inflammatoires locaux. La répétition des poussées est également responsable d’une destruction du cartilage, avec une déformation typique mais non spécifique « en pied de marmite » (
Phénotype trachéobronchique moins fréquent mais sévère
Cette forme concerne environ 25 % des patients ; elle est caractérisée par une chondrite de l’arbre trachéobronchique. Il s’agit de l’une des atteintes sévères de la maladie pouvant engager le pronostic vital. Ces formes posent une difficulté toute particulière, car le recours aux endoscopies ou à l’intubation (tableaux de détresse respiratoire aiguë) peut être responsable de perforations.
L’atteinte laryngée (40 % des patients) se manifeste par une toux sèche, une dysphonie (voire aphonie en cas d’œdème des cordes vocales). Parfois sont présents des cervicalgies antérieures aggravées par la palpation des structures cartilagineuses superficielles (pouvant faire évoquer à tort une thyroïdite subaiguë de De Quervain), une dyspnée laryngée, un stridor en cas de sténose sous-glottique.
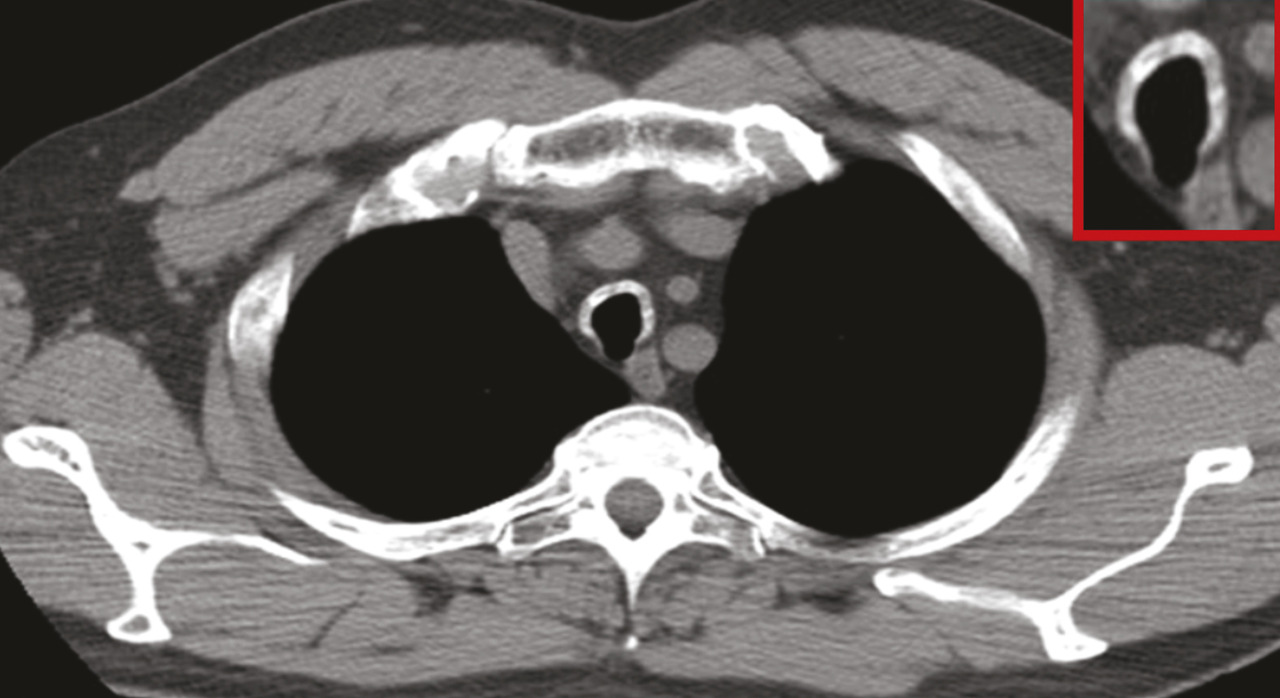
L’atteinte trachéobronchique est la plus grave et doit être dépistée systématiquement devant toute suspicion de PCA : elle est à évoquer devant une toux sèche, une dyspnée inhabituelle et parfois un wheezing. L’atteinte itérative menant à la destruction du cartilage trachéobronchique peut aboutir à un collapsus trachéobronchique et/ou à une sténose de la lumière trachéale, menant à des tableaux d’insuffisance respiratoire. Une particularité de cette atteinte est le respect habituel de la paroi postérieure de l’arbre trachéobronchique, qui est de nature membraneuse, sauf dans les formes évoluées où l’épaississement peut être circonférentiel (fig. 2 ). Les formes très évoluées peuvent nécessiter la mise en place de stents ou le recours à la chirurgie reconstructrice.
L’atteinte laryngée (40 % des patients) se manifeste par une toux sèche, une dysphonie (voire aphonie en cas d’œdème des cordes vocales). Parfois sont présents des cervicalgies antérieures aggravées par la palpation des structures cartilagineuses superficielles (pouvant faire évoquer à tort une thyroïdite subaiguë de De Quervain), une dyspnée laryngée, un stridor en cas de sténose sous-glottique.
L’atteinte trachéobronchique est la plus grave et doit être dépistée systématiquement devant toute suspicion de PCA : elle est à évoquer devant une toux sèche, une dyspnée inhabituelle et parfois un wheezing. L’atteinte itérative menant à la destruction du cartilage trachéobronchique peut aboutir à un collapsus trachéobronchique et/ou à une sténose de la lumière trachéale, menant à des tableaux d’insuffisance respiratoire. Une particularité de cette atteinte est le respect habituel de la paroi postérieure de l’arbre trachéobronchique, qui est de nature membraneuse, sauf dans les formes évoluées où l’épaississement peut être circonférentiel (
Phénotype associé au syndrome myélodysplasique : syndrome VEXAS
L’analyse des différents phénotypes présentés par les patients atteints de PCA avait mis en évidence un cluster phénotypique particulier, constitué par des hommes de plus de 60 ans ayant un syndrome myélodysplasique et une PCA de moins bon pronostic, souvent résistante aux traitements conventionnels.7 Fin 2020, une nouvelle entité nosologique, appelée syndrome VEXAS17, a décrit pour la première fois un syndrome auto-inflammatoire associé à une mutation somatique (et donc acquise) du gène UBA1 situé sur le chromosome X (ce qui explique la prédominance masculine) présente dans les cellules souches hématopoïétiques de la lignée myélomonocytaire. Cette découverte d’une première mutation acquise responsable d’un tableau auto-inflammatoire a révolutionné le monde de l’immunologie clinique : elle a permis de faire le lien entre différents tableaux cliniques encore incompris et de mauvais pronostic qui avaient déjà été identifiés dans le passé avec cette nouvelle entité, et notamment ce fameux troisième phénotype de PCA répondant moins bien aux thérapeutiques conventionnelles.
Ce type de PCA est à évoquer chez les hommes de plus de 50 ans présentant, de façon presque constante, une anémie macrocytaire avec un volume globulaire moyen (VGM) souvent supérieur à 110 fL. La fièvre et le syndrome inflammatoire biologique y sont plus fréquents, comme l’atteinte cutanée (à type de dermatose neutrophilique) et l’atteinte pulmonaire parenchymateuse. Des manifestations thromboemboliques sont également courantes. Il existe d’autres manifestations associées au syndrome VEXAS (la plus fréquente étant des tableaux de vascularite des gros vaisseaux type artérite à cellules géantes), et leur spectre ne cesse de s’étendre.19
Il existe aussi des PCA associées à des anomalies hématologiques sans mise en évidence de mutation UBA1 (probablement en lien avec d’autres mutations somatiques encore à découvrir).
Ce type de PCA est à évoquer chez les hommes de plus de 50 ans présentant, de façon presque constante, une anémie macrocytaire avec un volume globulaire moyen (VGM) souvent supérieur à 110 fL. La fièvre et le syndrome inflammatoire biologique y sont plus fréquents, comme l’atteinte cutanée (à type de dermatose neutrophilique) et l’atteinte pulmonaire parenchymateuse. Des manifestations thromboemboliques sont également courantes. Il existe d’autres manifestations associées au syndrome VEXAS (la plus fréquente étant des tableaux de vascularite des gros vaisseaux type artérite à cellules géantes), et leur spectre ne cesse de s’étendre.19
Il existe aussi des PCA associées à des anomalies hématologiques sans mise en évidence de mutation UBA1 (probablement en lien avec d’autres mutations somatiques encore à découvrir).
Autres atteintes plus rares de la PCA
Les autres atteintes de la PCA sont plus rares, résumées dans le tableau 2 .
La présence d’une atteinte neurologique (et notamment périphérique, exceptionnelle) ou d’une atteinte rénale doit faire évoquer systématiquement le diagnostic différentiel de vascularite à ANCA.
La présence d’une atteinte neurologique (et notamment périphérique, exceptionnelle) ou d’une atteinte rénale doit faire évoquer systématiquement le diagnostic différentiel de vascularite à ANCA.
Bilan initial devant toute suspicion de PCA
Le diagnostic de PCA ne peut être porté de façon formelle qu’en présence de chondrites. Le bilan initial recommandé est détaillé dans le tableau 3 .20 Il est bien sûr complété au besoin, en fonction des différentes manifestations cliniques (par exemple avis dermatologique et biopsie cutanée). Il sert à éliminer les principaux diagnostics différentiels, à identifier les maladies auto-immunes ou inflammatoires associées (jusqu’à 20 % des cas) [tableau 4 ] et à faire une cartographie initiale des atteintes présentes afin de procéder à une décision thérapeutique sur mesure et de programmer un suivi spécifique : réalisation d’une tomodensitométrie (TDM) cervicothoracique dynamique en inspiration et expiration, des explorations fonctionnelles respiratoires (EFR) avec mesure de la capacité de dif–fusion du monoxyde de carbone (DLCO) et test de marche des six minutes, un électrocardiogramme et une échocardiographie transthoracique. Le reste du bilan paraclinique est dicté par la présence d’éventuelles autres atteintes systémiques (neurosensorielles, dermatologiques…).
Il existe différents critères diagnostiques, dont les plus récents et les plus adaptés sont les critères de Michet.21 En pratique, ces critères ne sont néanmoins ni nécessaires ni suffisants pour porter le diagnostic de PCA et ne sont donc pas utilisés en pratique courante.
Il existe différents critères diagnostiques, dont les plus récents et les plus adaptés sont les critères de Michet.21 En pratique, ces critères ne sont néanmoins ni nécessaires ni suffisants pour porter le diagnostic de PCA et ne sont donc pas utilisés en pratique courante.
Amélioration du pronostic
Comme son nom l’indique, la PCA est une maladie chronique évoluant par poussées dont la fréquence et la gravité sont imprévisibles. Les séries les plus récentes mettent en évidence une franche amélioration du pronostic, avec des chiffres de survie à cinq et dix ans de 95 % et 91 % dans la série française de Cochin,7 et de 88 % et 81 % dans la série hongroise.4 Les facteurs de mauvais pronostic identifiés7 sont le sexe masculin, l’atteinte cardiaque et le phénotype associé au syndrome VEXAS ou aux autres hémopathies.18 L’importance des complications liées aux poussées itératives n’est pas négligeable : séquelles esthétiques, douleurs chroniques, troubles sensoriels, notamment surdité et baisse de l’acuité visuelle, troubles de l’équilibre, asthme persistant... Il est important de noter que même si le phénotype trachéobronchique est plus sévère que celui des chondrites mineures et nécessite une thérapeutique plus intensive, la mortalité n’y semble pas plus élevée.7 En revanche, le phénotype hématologique VEXAS possède un pronostic plus réservé, avec un taux de mortalité plus élevé.18
Traitements des poussées et parfois de fond
En raison de la rareté de la maladie et de l’absence d’essai randomisé, la prise en charge thérapeutique de la PCA n’est à l’heure actuelle pas formellement codifiée, même si le Protocole national de diagnostic et de soins (PNDS) constitue une première étape en ce sens20 et qu’un travail européen collaboratif a également permis de formuler des propositions thérapeutiques.8
Les principaux objectifs du traitement sont le contrôle des symptômes, la prévention des destructions cartilagineuses et des complications respiratoires et cardiovasculaires, la préservation de l’audition et de la vue, le maintien de la qualité de vie, et la prévention des effets indésirables des médicaments.20
Dans la majorité des cas, la PCA évolue par poussées, plus ou moins rapprochées dans le temps, de sévérité et de durée très variables et imprévisibles. La prise en charge de la maladie comprend donc le traitement des poussées qui peut être, selon les cas, purement symptomatique (anti-inflammatoires non stéroïdiens [AINS] et antalgiques) ou faire appel à une corticothérapie éventuellement associée aux immunosuppresseurs, et un traitement de fond (biothérapies...) chez certains patients, de façon à réduire la fréquence et la sévérité des poussées, à prévenir les séquelles, et/ou à réduire le besoin en corticoïdes.
Les biothérapies permettent de renforcer l’efficacité du traitement au cours des PCA graves ou corticorésistantes et pourraient être associées à une réduction de mortalité dans certaines séries récentes.22 L’infliximab a longtemps été utilisé en raison d’une efficacité chez environ la moitié des patients, mais avec de fréquentes pertes d’efficacité et des effets indésirables infectieux.23,24 L’étanercept semble également efficace.25 Plus récemment, l’efficacité du tocilizumab (anti-interleukine 6 [IL-6]) a été rapportée chez quelques patients, avec des atteintes sévères et réfractaires à de nombreuses lignes thérapeutiques.23,26-28 L’anakinra (IL-1RA) semble être efficace, avec quelques cas publiés.29,30 L’abatacept (analogue du CTLA-4) a également montré son efficacité en cas de résistance à trois anti-TNF et à un anti-IL-1.31 L’utilisation du rituximab semble quant à elle décevante, seule une étude sur neuf patients a montré une amélioration partielle chez deux patients sous ce traitement.32
Dans une revue récente de la littérature publiée par Arnaud et al.,33 les traitements les plus efficaces semblaient être l’abatacept (taux de réponse poolé de 72 %), le tocilizumab (66 %), les anti-TNF (64 %), l’anakinra (47 %) et le rituximab (43 %).
Les principaux objectifs du traitement sont le contrôle des symptômes, la prévention des destructions cartilagineuses et des complications respiratoires et cardiovasculaires, la préservation de l’audition et de la vue, le maintien de la qualité de vie, et la prévention des effets indésirables des médicaments.20
Dans la majorité des cas, la PCA évolue par poussées, plus ou moins rapprochées dans le temps, de sévérité et de durée très variables et imprévisibles. La prise en charge de la maladie comprend donc le traitement des poussées qui peut être, selon les cas, purement symptomatique (anti-inflammatoires non stéroïdiens [AINS] et antalgiques) ou faire appel à une corticothérapie éventuellement associée aux immunosuppresseurs, et un traitement de fond (biothérapies...) chez certains patients, de façon à réduire la fréquence et la sévérité des poussées, à prévenir les séquelles, et/ou à réduire le besoin en corticoïdes.
Les biothérapies permettent de renforcer l’efficacité du traitement au cours des PCA graves ou corticorésistantes et pourraient être associées à une réduction de mortalité dans certaines séries récentes.22 L’infliximab a longtemps été utilisé en raison d’une efficacité chez environ la moitié des patients, mais avec de fréquentes pertes d’efficacité et des effets indésirables infectieux.23,24 L’étanercept semble également efficace.25 Plus récemment, l’efficacité du tocilizumab (anti-interleukine 6 [IL-6]) a été rapportée chez quelques patients, avec des atteintes sévères et réfractaires à de nombreuses lignes thérapeutiques.23,26-28 L’anakinra (IL-1RA) semble être efficace, avec quelques cas publiés.29,30 L’abatacept (analogue du CTLA-4) a également montré son efficacité en cas de résistance à trois anti-TNF et à un anti-IL-1.31 L’utilisation du rituximab semble quant à elle décevante, seule une étude sur neuf patients a montré une amélioration partielle chez deux patients sous ce traitement.32
Dans une revue récente de la littérature publiée par Arnaud et al.,33 les traitements les plus efficaces semblaient être l’abatacept (taux de réponse poolé de 72 %), le tocilizumab (66 %), les anti-TNF (64 %), l’anakinra (47 %) et le rituximab (43 %).
Surveillance adaptée selon la gravité
En cas de poussée ou de forme grave, un suivi rapproché – tous les trois mois ou moins – semble légitime. Les examens paracliniques dépendent des atteintes présentées par le patient. L’activité de la maladie peut être évaluée par le relapsing polychondritis disease activity index (RPDAI)34 (tableau 5 ), et les complications par le relapsing polychondritis damage index (RPDAM)35 (tableau 6 ).
Trois phénotypes de pronostic et de prise en charge différents
La PCA est une pathologie chronique systémique évoluant par poussées imprévisibles, dont le diagnostic est essentiellement clinique et repose sur la présence de chondrites caractéristiques qui sont observées au début de la maladie uniquement dans un tiers des cas. Il existe trois principaux phénotypes de PCA imposant un traitement spécifique face à des pronostics différents. Il convient de rechercher de façon systématique l’atteinte trachéobronchique, responsable d’une grande partie de la morbi-mortalité de la maladie et pouvant mener le patient en réanimation pour détresse respiratoire aiguë. La recherche de syndrome VEXAS doit être systématique chez tout patient masculin de plus de 50 ans avec une anémie macrocytaire, d’autant plus s’il existe une atteinte dermatologique, parenchymateuse pulmonaire ou des complications thromboemboliques.
Le bilan initial permet d’éliminer les principaux diagnostics différentiels (notamment les vascularites à ANCA) et de rechercher une maladie inflammatoire associée dans 30 % des cas. Les facteurs de mauvais pronostic sont le sexe masculin, la présence d’une atteinte cardiovasculaire et le syndrome VEXAS. La prise en charge thérapeutique de la PCA reste encore mal codifiée.
Le bilan initial permet d’éliminer les principaux diagnostics différentiels (notamment les vascularites à ANCA) et de rechercher une maladie inflammatoire associée dans 30 % des cas. Les facteurs de mauvais pronostic sont le sexe masculin, la présence d’une atteinte cardiovasculaire et le syndrome VEXAS. La prise en charge thérapeutique de la PCA reste encore mal codifiée.
Références
1. Bandeira M, Di Cianni F, Marinello D, et al. An overlook on the current registries for rare and complex connective tissue diseases and the future scenario of TogethERN ReCONNET. Front Med 2022;9:889997.
2. Talarico R, Aguilera S, Alexander T, et al. The added value of a European Reference Network on rare and complex connective tissue and musculoskeletal diseases: Insights after the first 5 years of the ERN ReCONNET. Clin Exp Rheumatol 2022;40 Suppl 134(5):3‑11.
3. Hazra N, Dregan A, Charlton J, Gulliford MC, D’Cruz DP. Incidence and mortality of relapsing polychondritis in the UK: A population-based cohort study. Rheumatol Oxf Engl 2015;54(12):2181‑7.
4. Horváth A, Páll N, Molnár K, et al. A nationwide study of the epidemiology of relapsing polychondritis. Clin Epidemiol 2016;8:211‑30.
5. Kent PD, Michet CJ, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol 2004;16(1):56‑61.
6. Chen N, Zheng Y. Characteristics and clinical outcomes of 295 patients with relapsing polychondritis. J Rheumatol 2021;48(12):1876‑82.
7. Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: Analysis of a recent series of 142 patients. Arthritis Rheumatol Hoboken NJ 2016;68(12):2992‑3001.
8. Belot A, Duquesne A, Job-Deslandre C, et al. Pediatric-onset relapsing polychondritis: Case series and systematic review. J Pediatr 2010;156(3):484‑9.
9. Alqanatish JT, Alshanwani JR. Relapsing polychondritis in children: A review. Mod Rheumatol 2020;30(5):788‑98.
10. Shimizu J, Yamano Y, Kawahata K, Suzuki N. Nationwide cross-sectional survey of patients with relapsing polychondritis in 2019 demonstrates reduction of airway involvement compared with that in 2009. Sci Rep 2022;12(1):465.
11. Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: An autoimmune disease with many faces. Autoimmun Rev 2010;9(8):540‑6.
12. Buckner JH, Van Landeghen M, Kwok WW, Tsarknaridis L. Identification of type II collagen peptide 261-273-specific T cell clones in a patient with relapsing polychondritis. Arthritis Rheum 2002;46(1):238‑44.
13. Stabler T, Piette JC, Chevalier X, Marini-Portugal A, Kraus VB. Serum cytokine profiles in relapsing polychondritis suggest monocyte/macrophage activation. Arthritis Rheum2004;50(11):3663‑7.
14. Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med 1978;299(22):1203‑7.
15. Ebringer R, Rook G, Swana GT, Bottazzo GF, Doniach D. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis 1981;40(5):473‑9.
16. Hansson AS, Heinegård D, Holmdahl R. A new animal model for relapsing polychondritis, induced by cartilage matrix protein (matrilin-1). J Clin Invest 1999;104(5):589‑98.
17. Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020;383(27):2628‑38.
18. Khitri MY, Guedon AF, Georgin-Lavialle S, et al. Comparison between idiopathic and VEXAS-relapsing polychondritis: Analysis of a French case series of 95 patients. RMD Open 2022;8(2):e002255.
19. Al-Hakim A, Savic S. An update on VEXAS syndrome. Expert Rev Clin Immunol 2023;19(2):203-15.
20. Polychondrite chronique atrophiante [internet]. Haute Autorité de santé. https://vu.fr/PfDN
21. Michet CJ, McKenna CH, Luthra HS, O’Fallon WM. Relapsing polychondritis. Survival and predictive role of early disease manifestations. Ann Intern Med 1986;104(1):74‑8.
22. Shimizu J, Yamano Y, Kawahata K, Suzuki N. Nationwide cross-sectional survey of patients with relapsing polychondritis in 2019 demonstrates reduction of airway involvement compared with that in 2009. Sci Rep 2022;12(1):465.
23. Moulis G, Sailler L, Pugnet G, Astudillo L, Arlet P. Biologics in relapsing polychondritis: A case series. Clin Exp Rheumatol 2013;31(6):937-9.
24. Kemta Lepka F, Kraus VB, Chevalier X. Biologics in relapsing polychondritis: A literature review. Semin Arthritis Rheum 2012;41(5):712-9.
25. Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol 2005;32(7):1413.
26. Stael E, Smith V, Wittoek R, Creytens D, Mielants. Sustained response to tocilizumab in a patient with relapsing polychondritis with aortic involvement: A case based review. Clin Rheumato 2015;34(1):189-93.
27. Farhat R, Clavel G, Villeneuve D, et al. Sustained remission with tocilizumab in refractory relapsing polychondritis with ocular involvement: A case series. Ocul Immunol Inflamm 2021;29(1):9-13.
28. Liao HT. Efficacy of tocilizumab for refractory relapsing polychondritis with tracheal stenosis and respiratory distress. Rheumatology (Oxford) 2022;61(3):1293-4.
29. Vounotrypidis P, Sakellariou GT, Zisopoulos D, Berberidis C. Refractory relapsing polychondritis: Rapid and sustained response in the treatment with an IL-1 receptor antagonist (anakinra). Rheumatology (Oxford) 2006;45(4):491-2.
30. Wendling D, Govindaraju S, Prati C, Toussirot E, Bertolini E. Efficacy of anakinra in a patient with refractory relapsing polychondritis. Joint Bone Spine 2008;75(5):622-4.
31. Moulis G, Pugnet G, Sailler L, Astudillo L, Arlet P. Abatacept in relapsing polychondritis. Ann Rheum Dis 2013;72(11):e27.
32. Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: A retrospective study of nine patients. Arthritis Rheum 2009;61(5):577-82.
33. Petitdemange A, Sztejkowski C, Damian L, et al. Treatment of relapsing polychondritis: A systematic review. Clin Exp Rheumatol 2022;40 Suppl 134(5):81-5.
34. Arnaud L, Devilliers H, Peng SL, et al. The relapsing polychondritis disease activity index: Development of a disease activity score for relapsing polychondritis. Autoimmun Rev 2012;12(2):204‑9.
35. Mertz P, Belot A, Cervera R, et al. The relapsing polychondritis damage index (RPDAM): Development of a disease-specific damage score for relapsing polychondritis. Jt Bone Spine Rev Rhum 2019;86(3):363-8.
2. Talarico R, Aguilera S, Alexander T, et al. The added value of a European Reference Network on rare and complex connective tissue and musculoskeletal diseases: Insights after the first 5 years of the ERN ReCONNET. Clin Exp Rheumatol 2022;40 Suppl 134(5):3‑11.
3. Hazra N, Dregan A, Charlton J, Gulliford MC, D’Cruz DP. Incidence and mortality of relapsing polychondritis in the UK: A population-based cohort study. Rheumatol Oxf Engl 2015;54(12):2181‑7.
4. Horváth A, Páll N, Molnár K, et al. A nationwide study of the epidemiology of relapsing polychondritis. Clin Epidemiol 2016;8:211‑30.
5. Kent PD, Michet CJ, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol 2004;16(1):56‑61.
6. Chen N, Zheng Y. Characteristics and clinical outcomes of 295 patients with relapsing polychondritis. J Rheumatol 2021;48(12):1876‑82.
7. Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: Analysis of a recent series of 142 patients. Arthritis Rheumatol Hoboken NJ 2016;68(12):2992‑3001.
8. Belot A, Duquesne A, Job-Deslandre C, et al. Pediatric-onset relapsing polychondritis: Case series and systematic review. J Pediatr 2010;156(3):484‑9.
9. Alqanatish JT, Alshanwani JR. Relapsing polychondritis in children: A review. Mod Rheumatol 2020;30(5):788‑98.
10. Shimizu J, Yamano Y, Kawahata K, Suzuki N. Nationwide cross-sectional survey of patients with relapsing polychondritis in 2019 demonstrates reduction of airway involvement compared with that in 2009. Sci Rep 2022;12(1):465.
11. Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: An autoimmune disease with many faces. Autoimmun Rev 2010;9(8):540‑6.
12. Buckner JH, Van Landeghen M, Kwok WW, Tsarknaridis L. Identification of type II collagen peptide 261-273-specific T cell clones in a patient with relapsing polychondritis. Arthritis Rheum 2002;46(1):238‑44.
13. Stabler T, Piette JC, Chevalier X, Marini-Portugal A, Kraus VB. Serum cytokine profiles in relapsing polychondritis suggest monocyte/macrophage activation. Arthritis Rheum2004;50(11):3663‑7.
14. Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med 1978;299(22):1203‑7.
15. Ebringer R, Rook G, Swana GT, Bottazzo GF, Doniach D. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis 1981;40(5):473‑9.
16. Hansson AS, Heinegård D, Holmdahl R. A new animal model for relapsing polychondritis, induced by cartilage matrix protein (matrilin-1). J Clin Invest 1999;104(5):589‑98.
17. Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020;383(27):2628‑38.
18. Khitri MY, Guedon AF, Georgin-Lavialle S, et al. Comparison between idiopathic and VEXAS-relapsing polychondritis: Analysis of a French case series of 95 patients. RMD Open 2022;8(2):e002255.
19. Al-Hakim A, Savic S. An update on VEXAS syndrome. Expert Rev Clin Immunol 2023;19(2):203-15.
20. Polychondrite chronique atrophiante [internet]. Haute Autorité de santé. https://vu.fr/PfDN
21. Michet CJ, McKenna CH, Luthra HS, O’Fallon WM. Relapsing polychondritis. Survival and predictive role of early disease manifestations. Ann Intern Med 1986;104(1):74‑8.
22. Shimizu J, Yamano Y, Kawahata K, Suzuki N. Nationwide cross-sectional survey of patients with relapsing polychondritis in 2019 demonstrates reduction of airway involvement compared with that in 2009. Sci Rep 2022;12(1):465.
23. Moulis G, Sailler L, Pugnet G, Astudillo L, Arlet P. Biologics in relapsing polychondritis: A case series. Clin Exp Rheumatol 2013;31(6):937-9.
24. Kemta Lepka F, Kraus VB, Chevalier X. Biologics in relapsing polychondritis: A literature review. Semin Arthritis Rheum 2012;41(5):712-9.
25. Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol 2005;32(7):1413.
26. Stael E, Smith V, Wittoek R, Creytens D, Mielants. Sustained response to tocilizumab in a patient with relapsing polychondritis with aortic involvement: A case based review. Clin Rheumato 2015;34(1):189-93.
27. Farhat R, Clavel G, Villeneuve D, et al. Sustained remission with tocilizumab in refractory relapsing polychondritis with ocular involvement: A case series. Ocul Immunol Inflamm 2021;29(1):9-13.
28. Liao HT. Efficacy of tocilizumab for refractory relapsing polychondritis with tracheal stenosis and respiratory distress. Rheumatology (Oxford) 2022;61(3):1293-4.
29. Vounotrypidis P, Sakellariou GT, Zisopoulos D, Berberidis C. Refractory relapsing polychondritis: Rapid and sustained response in the treatment with an IL-1 receptor antagonist (anakinra). Rheumatology (Oxford) 2006;45(4):491-2.
30. Wendling D, Govindaraju S, Prati C, Toussirot E, Bertolini E. Efficacy of anakinra in a patient with refractory relapsing polychondritis. Joint Bone Spine 2008;75(5):622-4.
31. Moulis G, Pugnet G, Sailler L, Astudillo L, Arlet P. Abatacept in relapsing polychondritis. Ann Rheum Dis 2013;72(11):e27.
32. Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: A retrospective study of nine patients. Arthritis Rheum 2009;61(5):577-82.
33. Petitdemange A, Sztejkowski C, Damian L, et al. Treatment of relapsing polychondritis: A systematic review. Clin Exp Rheumatol 2022;40 Suppl 134(5):81-5.
34. Arnaud L, Devilliers H, Peng SL, et al. The relapsing polychondritis disease activity index: Development of a disease activity score for relapsing polychondritis. Autoimmun Rev 2012;12(2):204‑9.
35. Mertz P, Belot A, Cervera R, et al. The relapsing polychondritis damage index (RPDAM): Development of a disease-specific damage score for relapsing polychondritis. Jt Bone Spine Rev Rhum 2019;86(3):363-8.
Dans cet article
- Maladie rare, mal répertoriée
- À la frontière de l’immunité innée et adaptative
- Principaux phénotypes cliniques de PCA
- Autres atteintes plus rares de la PCA
- Bilan initial devant toute suspicion de PCA
- Amélioration du pronostic
- Traitements des poussées et parfois de fond
- Surveillance adaptée selon la gravité
- Trois phénotypes de pronostic et de prise en charge différents

Résumé
La polychondrite chronique atrophiante (PCA) est une maladie systémique évoluant par poussées imprévisibles, dont le diagnostic est essentiellement clinique et repose sur l’existence de chondrites caractéristiques qui ne sont présentes au début de la maladie que dans un tiers des cas. Trois principaux phénotypes de PCA sont décrits, imposant une prise en charge thérapeutique spécifique face à des pronostics différents. Il convient de rechercher de façon systématique l’atteinte trachéobronchique, responsable d’une grande partie de la morbi-mortalité de la maladie. Il en est de même pour le syndrome VEXAS (Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) mais uniquement chez tout patient masculin de plus de 50 ans avec une anémie macrocytaire, d’autant plus s’il existe une atteinte dermatologique, parenchymateuse pulmonaire ou des complications thromboemboliques. Le bilan initial permet d’éliminer les principaux diagnostics différentiels (notamment les vascularites à anticorps anticytoplasme des polynucléaires neutrophiles [ANCA]) et de rechercher une maladie associée (auto-immune ou inflammatoire) dans 30 % des cas. La prise en charge thérapeutique de la PCA reste encore mal codifiée et dépend de sa gravité.