Bien que rares, les tumeurs stromales gastro-intestinales (GIST) correspondent au sous-type histologique de sarcomes le plus fréquent. Elles constituent un modèle emblématique du succès des thérapies ciblées en oncologie. La découverte des mutations des gènes KIT et PDGFRA et la réponse aux inhibiteurs de tyrosine kinase des différents sous-types de GIST a permis la mise en place de stratégies thérapeutiques personnalisées et une augmentation majeure de la survie.
Les tumeurs stromales gastro-intestinales (GIST) constituent le sous-type histologique de sarcomes le plus fréquent. L’incidence annuelle est d’environ 12 cas par million d’individus, ce qui représente environ 800 cas par an en France.1 Les GIST sont issues de la transformation tumorale des cellules interstitielles de Cajal qui sont les cellules responsables de la contractilité du tube digestif. Les localisations les plus fréquentes des GIST sont l’estomac, l’intestin grêle et le rectum. Les récidives métastatiques concernent le plus souvent le foie et le péritoine. Les GIST représentent un des modèles les plus emblématiques du succès des thérapies ciblées en oncologie. En effet, avant la découverte des bases moléculaires de l’oncogenèse de ces tumeurs, le pronostic des patients au stade avancé était très réservé du fait de l’absence d’efficacité des chimiothérapies conventionnelles. La mise en évidence de mutations activatrices des gènes KIT et PDGFRA dans la majorité des GIST a été le rationnel pour le développement de thérapies ciblées qui ont transformé la prise en charge des patients et ont permis une augmentation de la survie globale au stade métastatique de moins de dix-huit mois il y a une vingtaine d’années à plus de soixante-dix mois aujourd’hui.2
Essentielle détection des anomalies moléculaires lors du diagnostic
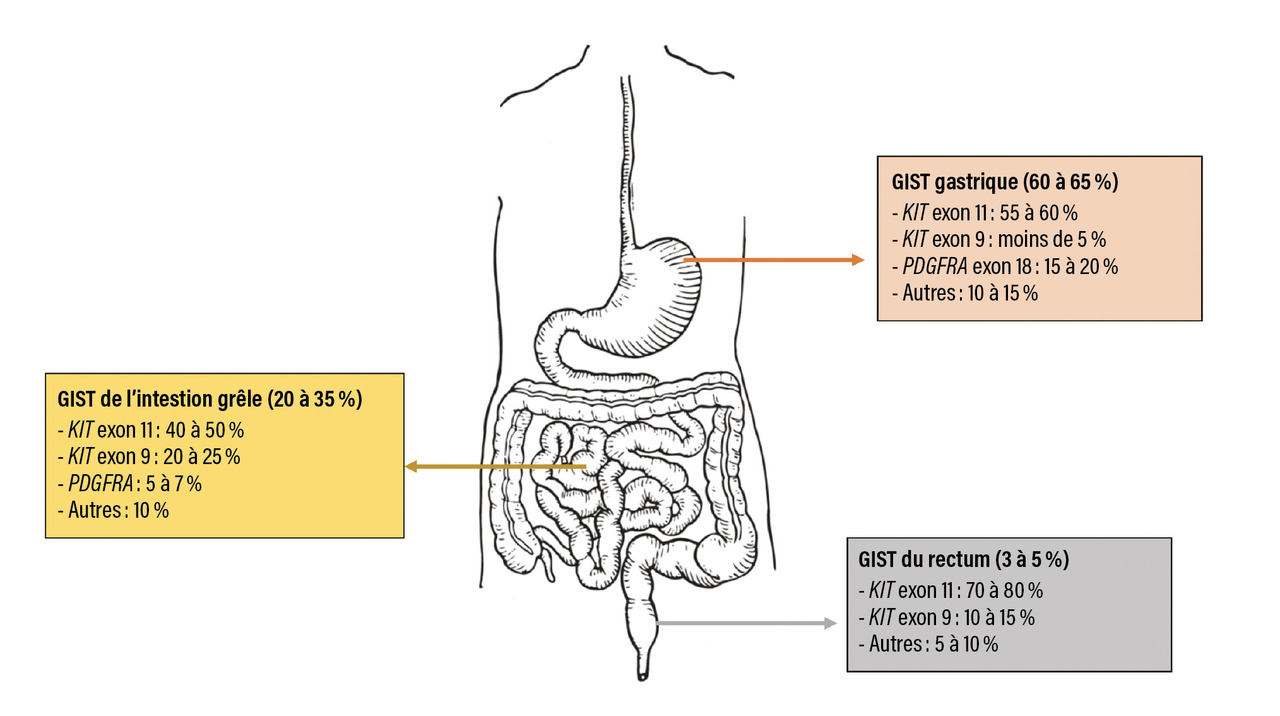
L’immense majorité des GIST se développe de façon sporadique, sans facteur de risque reconnu. Le développement des GIST est associé à des anomalies moléculaires oncogéniques, les plus fréquentes étant les mutations activatrices dans les gènes KIT (60 à 70 % des cas) et PDGFRA (10 à 15 % des cas).3,4 Les gènes KIT et PDGFRA codent pour des récepteurs à tyrosine kinase (RTK) homologues dont l’activation incontrôlée conduit à une stimulation des voies de signalisation impliquées dans la prolifération cellulaire responsables de la transformation en cellules tumorales. Plus de 150 mutations de KIT et PDGFRA ont été décrites, mais les plus fréquentes sont localisées au niveau des exons 11 et 9 du gène KIT et au niveau de l’exon 18 de PDGFRA. Il existe une corrélation entre la localisation de la tumeur primitive et le génotype : les mutations KIT exon 11 et PDGFRA exon 18, par exemple, sont plus fréquentes au niveau de l’estomac, tandis que les mutations KIT exon 9 sont plus répandues dans les tumeurs de l’intestin grêle (figure ).
Dix à quinze pour cent des GIST ne présentent pas de mutation de KIT ni de PDGFRA mais sont caractérisées par des anomalies moléculaires plus rares telles que :
La détection initiale des anomalies moléculaires est importante à la fois pour le diagnostic et pour les décisions thérapeutiques car le gène affecté et le type de mutation conditionnent le pronostic de la maladie et la sensibilité aux traitements médicaux (tableau 1 ).
Dix à quinze pour cent des GIST ne présentent pas de mutation de KIT ni de PDGFRA mais sont caractérisées par des anomalies moléculaires plus rares telles que :
- des inactivations des gènes SDH ou NF1 ;
- des mutations activatrices de BRAF ;
- ou des gènes de fusion impliquant d’autres RTK.2
La détection initiale des anomalies moléculaires est importante à la fois pour le diagnostic et pour les décisions thérapeutiques car le gène affecté et le type de mutation conditionnent le pronostic de la maladie et la sensibilité aux traitements médicaux (
Imagerie et biopsie pour confirmer le diagnostic
La présentation clinique des GIST est le plus souvent similaire à celle d’un autre type de cancer gastro-intestinal. Une hémorragie digestive, des douleurs abdominales, une augmentation de volume de l’abdomen ou des symptômes occlusifs sont possibles, en fonction de la localisation. Les GIST de petite taille (moins de 2 cm) sont le plus souvent découvertes de façon fortuite lors d’endoscopies ou de gestes chirurgicaux réalisés pour une autre indication et peuvent bénéficier d’une simple surveillance endoscopique en cas de localisation gastrique.
En cas de découverte de GIST, le bilan d’extension initial doit reposer sur la réalisation d’une tomodensitométrie thoraco-abdomino-pelvienne (TDM-TAP) avec injection de produit de contraste.5 L’imagerie par résonance magnétique (IRM) peut être utile en cas de GIST du rectum et la tomographie par émission de positons (TEP) au 18FDG en cas de doute sur une lésion métastatique au scanner ou à l’IRM.
Si la tumeur est opérable, une biopsie préopératoire doit être discutée en réunion de concertation pluridisciplinaire (RCP) spécialisée ; elle est recommandée mais non indispensable en cas de très forte suspicion diagnostique lorsqu’une chirurgie simple et non mutilante est possible. La biopsie est, en revanche, indispensable en cas de doute diagnostique avec une autre tumeur ou de maladie avancée ou métastasique relevant d’un traitement médical initial. La biopsie peut porter sur la tumeur primitive ou sur une métastase, et être réalisée sous écho-endoscopie ou par voie percutanée. Elle doit, dans tous les cas, être de taille suffisante pour permettre les analyses histologiques et moléculaires.
Le diagnostic de GIST est porté sur l’analyse histologique standard ; il s’agit de tumeurs malignes à cellules fusiformes exprimant dans 95 % des cas les marqueurs KIT (CD117) et DOG-1 en immunohistochimie. La relecture des lames en centre expert de référence est recommandée pour tous les sarcomes, dont les GIST (Réseau de référence en pathologie des sarcomes, RRePS). La recherche de mutations des gènes KIT et PDGFRA doit être réalisée par une technique de biologie moléculaire. En l’absence de mutation KIT ou PDGFRA, les autres sous-types moléculaires plus rares doivent être recherchés.
En cas de découverte de GIST, le bilan d’extension initial doit reposer sur la réalisation d’une tomodensitométrie thoraco-abdomino-pelvienne (TDM-TAP) avec injection de produit de contraste.5 L’imagerie par résonance magnétique (IRM) peut être utile en cas de GIST du rectum et la tomographie par émission de positons (TEP) au 18FDG en cas de doute sur une lésion métastatique au scanner ou à l’IRM.
Si la tumeur est opérable, une biopsie préopératoire doit être discutée en réunion de concertation pluridisciplinaire (RCP) spécialisée ; elle est recommandée mais non indispensable en cas de très forte suspicion diagnostique lorsqu’une chirurgie simple et non mutilante est possible. La biopsie est, en revanche, indispensable en cas de doute diagnostique avec une autre tumeur ou de maladie avancée ou métastasique relevant d’un traitement médical initial. La biopsie peut porter sur la tumeur primitive ou sur une métastase, et être réalisée sous écho-endoscopie ou par voie percutanée. Elle doit, dans tous les cas, être de taille suffisante pour permettre les analyses histologiques et moléculaires.
Le diagnostic de GIST est porté sur l’analyse histologique standard ; il s’agit de tumeurs malignes à cellules fusiformes exprimant dans 95 % des cas les marqueurs KIT (CD117) et DOG-1 en immunohistochimie. La relecture des lames en centre expert de référence est recommandée pour tous les sarcomes, dont les GIST (Réseau de référence en pathologie des sarcomes, RRePS). La recherche de mutations des gènes KIT et PDGFRA doit être réalisée par une technique de biologie moléculaire. En l’absence de mutation KIT ou PDGFRA, les autres sous-types moléculaires plus rares doivent être recherchés.
Prise en charge thérapeutique par les centres experts
La prise en charge thérapeutique des patients atteints de GIST doit être discutée en RCP de centre expert pour la prise en charge des sarcomes (réseau de référence NetSarc+ labellisé par l’Institut national du cancer [INCa] comportant 26 centres spécialisés sur le territoire national).5
Exérèse chirurgicale curative en cas de maladie localisée et résécable
L’exérèse chirurgicale complète de la tumeur en monobloc est le seul traitement curatif des GIST et doit être proposée en première intention en cas de maladie localisée et résécable. Elle nécessite une expertise chirurgicale car les GIST sont des tumeurs fragiles et une rupture tumorale peropératoire est associée de manière certaine à une récidive métastatique péritonéale. Après résection chirurgicale, l’estimation du risque de récidive est primordiale pour décider de l’indication d’un traitement adjuvant (tableau 2 ). Plusieurs classifications existent ; les plus utilisées étant la classification AFIP (Armed Forces Institute of Pathology) de Miettinen et la classification NIH (National Institutes of Health) modifiée par Joensuu, qui sont fondées sur trois critères : la taille tumorale, la localisation de la tumeur et l’indice mitotique.6
La chirurgie en situation métastatique ne doit pas être le traitement de première intention, et doit être discutée au cas par cas en centre expert, en tenant compte du risque de complications per- et postopératoires.
La chirurgie en situation métastatique ne doit pas être le traitement de première intention, et doit être discutée au cas par cas en centre expert, en tenant compte du risque de complications per- et postopératoires.
Traitement médical fondé sur les inhibiteurs de tyrosine kinase
Les GIST sont des maladies chimiorésistantes, et la chimiothérapie cytotoxique n’est pas indiquée en dehors de situations exceptionnelles.
Depuis la découverte des mutations KIT et PDGFRA il y a une vingtaine d’années et la démonstration de l’efficacité des thérapies ciblées ciblant ces mutations, les inhibiteurs de tyrosine kinase (ITK) constituent la pierre angulaire du traitement médical des GIST.
L’imatinib est un ITK initialement développé pour le traitement de la leucémie myéloïde chronique qui inhibe également les protéines kinases KIT et PDGFRA. L’imatinib est le traitement de première ligne pour les GIST avancées ; il est également indiqué en situation préopératoire (néoadjuvante) dans les GIST de résécabilité limite et en situation postopératoire (adjuvante) dans les GIST à haut risque de récidive.5 La dose standard est de 400 mg par jour par voie orale, en continu.
Le traitement est généralement très bien toléré, les principaux effets indésirables étant des œdèmes périorbitaires, des nausées, des diarrhées et des rashs qui nécessitent rarement des adaptations posologiques. Une vigilance particulière doit être portée sur les traitements concomitants car il existe un risque d’interactions médicamenteuses pouvant conduire à une mauvaise tolérance liée à une surexposition plasmatique ou, à l’inverse, à une sous-exposition au traitement. Des dosages plasmatiques de la molécule peuvent aider à adapter la dose chez les patients tolérant mal le traitement ou en cas d’absence d’efficacité.
En situation adjuvante pour les tumeurs à haut risque de récidive, l’imatinib doit être réservé aux GIST avec mutations KIT et PDGFRA sensibles au traitement.7 La durée totale de traitement est de trois ans. Un traitement adjuvant dans les autres sous-types moléculaires n’est pas indiqué.
En situation métastatique, l’imatinib conduit à des réponses tumorales chez la majorité des patients. La durée de contrôle de la maladie atteint généralement plus de deux ans mais peut varier en fonction des sous-types moléculaires.8 La progression après une réponse initiale sous imatinib témoigne de l’émergence de mutations de résistance au traitement et nécessite l’utilisation d’autres ITK. Les GIST ne présentant pas de mutation KIT ou PDGFRA ou présentant une mutation particulière de PDGFRA (D842V) sont résistantes d’emblée à l’imatinib.
Après progression sous imatinib, le traitement repose sur l’utilisation successive d’autres ITK de spectre d’action plus large que l’imatinib : respectivement le sunitinib en deuxième ligne, le régorafénib en troisième ligne et le riprétinib en quatrième ligne (tableau 3 ). Ces ITK sont associés à des taux de réponse et à des durées de contrôle de la maladie plus faibles que l’imatinib, avec des profils de tolérance moins favorables, en particulier pour le sunitinib et le régorafénib.9-11 Là encore, l’efficacité du traitement dépend du sous-type moléculaire initial et du type de mutation de résistance développé sous imatinib. Au-delà de la quatrième ligne de traitement, une réintroduction de l’imatinib est possible, et l’orientation des patients vers des essais cliniques est recommandée.
Pour les patients ayant des formes moléculaires d’emblée résistantes à l’imatinib (résistance primaire), plusieurs molécules ont montré leur bénéfice majeur : l’avapritinib pour les GIST avec mutation PDGFRA D842V, le larotrectinib et l’entrectinib pour les GIST avec fusion NTRK.12 Le traitement optimal d’autres sous-types rares (comme les GIST avec mutations NF1 ou SDH) reste inconnu.
Depuis la découverte des mutations KIT et PDGFRA il y a une vingtaine d’années et la démonstration de l’efficacité des thérapies ciblées ciblant ces mutations, les inhibiteurs de tyrosine kinase (ITK) constituent la pierre angulaire du traitement médical des GIST.
L’imatinib est un ITK initialement développé pour le traitement de la leucémie myéloïde chronique qui inhibe également les protéines kinases KIT et PDGFRA. L’imatinib est le traitement de première ligne pour les GIST avancées ; il est également indiqué en situation préopératoire (néoadjuvante) dans les GIST de résécabilité limite et en situation postopératoire (adjuvante) dans les GIST à haut risque de récidive.5 La dose standard est de 400 mg par jour par voie orale, en continu.
Le traitement est généralement très bien toléré, les principaux effets indésirables étant des œdèmes périorbitaires, des nausées, des diarrhées et des rashs qui nécessitent rarement des adaptations posologiques. Une vigilance particulière doit être portée sur les traitements concomitants car il existe un risque d’interactions médicamenteuses pouvant conduire à une mauvaise tolérance liée à une surexposition plasmatique ou, à l’inverse, à une sous-exposition au traitement. Des dosages plasmatiques de la molécule peuvent aider à adapter la dose chez les patients tolérant mal le traitement ou en cas d’absence d’efficacité.
En situation adjuvante pour les tumeurs à haut risque de récidive, l’imatinib doit être réservé aux GIST avec mutations KIT et PDGFRA sensibles au traitement.7 La durée totale de traitement est de trois ans. Un traitement adjuvant dans les autres sous-types moléculaires n’est pas indiqué.
En situation métastatique, l’imatinib conduit à des réponses tumorales chez la majorité des patients. La durée de contrôle de la maladie atteint généralement plus de deux ans mais peut varier en fonction des sous-types moléculaires.8 La progression après une réponse initiale sous imatinib témoigne de l’émergence de mutations de résistance au traitement et nécessite l’utilisation d’autres ITK. Les GIST ne présentant pas de mutation KIT ou PDGFRA ou présentant une mutation particulière de PDGFRA (D842V) sont résistantes d’emblée à l’imatinib.
Après progression sous imatinib, le traitement repose sur l’utilisation successive d’autres ITK de spectre d’action plus large que l’imatinib : respectivement le sunitinib en deuxième ligne, le régorafénib en troisième ligne et le riprétinib en quatrième ligne (
Pour les patients ayant des formes moléculaires d’emblée résistantes à l’imatinib (résistance primaire), plusieurs molécules ont montré leur bénéfice majeur : l’avapritinib pour les GIST avec mutation PDGFRA D842V, le larotrectinib et l’entrectinib pour les GIST avec fusion NTRK.12 Le traitement optimal d’autres sous-types rares (comme les GIST avec mutations NF1 ou SDH) reste inconnu.
Modalités de suivi sous traitement médical et surveillance
Le suivi des patients sous ITK comporte généralement un examen clinique et un bilan biologique mensuel pour s’assurer de la tolérance du traitement, évaluer l’observance et détecter des signes cliniques de réponse ou de progression tumorale.
Le suivi radiologique se fait tous les trois mois et repose principalement sur l’utilisation de la TDM TAP avec injection de produit de contraste. Les critères classiques de réponse tumorale fondés sur la mesure des cibles ne sont pas bien adaptés aux GIST. En effet, en cas de réponse sous ITK, la masse tumorale devient hypodense et les prises de contraste régressent rapidement tandis que la diminution de taille est plus lente et inconstante. Des critères radiologiques dédiés à l’évaluation de la réponse des GIST sous ITK ont été développés, ils sont fondés sur la densité tumorale et doivent être utilisés en première intention (critères de Choi).13
Pour les patients ayant bénéficié d’un traitement à visée curative, une surveillance trimestrielle pendant trois ans puis semestrielle pendant deux ans supplémentaires doit être réalisée, comportant au minimum un examen clinique, un bilan biologique complet et une imagerie abdomino-pelvienne (TDM ou IRM). Les rechutes au-delà de cinq ans sont très rares.
Le suivi radiologique se fait tous les trois mois et repose principalement sur l’utilisation de la TDM TAP avec injection de produit de contraste. Les critères classiques de réponse tumorale fondés sur la mesure des cibles ne sont pas bien adaptés aux GIST. En effet, en cas de réponse sous ITK, la masse tumorale devient hypodense et les prises de contraste régressent rapidement tandis que la diminution de taille est plus lente et inconstante. Des critères radiologiques dédiés à l’évaluation de la réponse des GIST sous ITK ont été développés, ils sont fondés sur la densité tumorale et doivent être utilisés en première intention (critères de Choi).13
Pour les patients ayant bénéficié d’un traitement à visée curative, une surveillance trimestrielle pendant trois ans puis semestrielle pendant deux ans supplémentaires doit être réalisée, comportant au minimum un examen clinique, un bilan biologique complet et une imagerie abdomino-pelvienne (TDM ou IRM). Les rechutes au-delà de cinq ans sont très rares.
Génotypage en routine pour une stratégie thérapeutique personnalisée
Les GIST représentent un bon exemple des bénéfices de la médecine personnalisée en oncologie. La découverte des mutations des gènes KIT et PDGFRA, puis le démembrement moléculaire des formes associées à des mutations de résistance primaire ou secondaire aux ITK ont conduit à la mise en place de stratégies thérapeutiques de plus en plus personnalisées et à une augmentation majeure de la survie, particulièrement en situation avancée, qui est aujourd’hui de plus de six ans.
L’histoire naturelle et la réponse aux ITK des différents sous-types de GIST sont désormais bien connues, mais plusieurs questions restent en suspens concernant la prise en charge thérapeutique en routine dans certaines situations, en particulier la prise en charge thérapeutique optimale des GIST de taille inférieure à 2 cm, la place optimale de la chirurgie en situation métastatique et le traitement médical de certains sous-types moléculaires très rares.
De nouveaux ITK sont d’ores et déjà en développement et leur positionnement dans la stratégie médicale sera la question des futurs essais cliniques. La détection des mutations de résistance secondaire, grâce à l’utilisation de biomarqueurs sanguins comme l’ADN tumoral circulant, déterminera probablement la stratégie d’utilisation des ITK dans un futur très proche. Dans tous les cas, les patients atteints de GIST doivent aujourd’hui bénéficier en routine du génotypage de leur maladie pour la prise en charge la plus adaptée. L’orientation des patients vers des centres de référence, particulièrement en cas de maladie avancée, leur permet de bénéficier de cette prise en charge optimale et de se voir possiblement proposer l’inclusion dans des essais cliniques innovants.
L’histoire naturelle et la réponse aux ITK des différents sous-types de GIST sont désormais bien connues, mais plusieurs questions restent en suspens concernant la prise en charge thérapeutique en routine dans certaines situations, en particulier la prise en charge thérapeutique optimale des GIST de taille inférieure à 2 cm, la place optimale de la chirurgie en situation métastatique et le traitement médical de certains sous-types moléculaires très rares.
De nouveaux ITK sont d’ores et déjà en développement et leur positionnement dans la stratégie médicale sera la question des futurs essais cliniques. La détection des mutations de résistance secondaire, grâce à l’utilisation de biomarqueurs sanguins comme l’ADN tumoral circulant, déterminera probablement la stratégie d’utilisation des ITK dans un futur très proche. Dans tous les cas, les patients atteints de GIST doivent aujourd’hui bénéficier en routine du génotypage de leur maladie pour la prise en charge la plus adaptée. L’orientation des patients vers des centres de référence, particulièrement en cas de maladie avancée, leur permet de bénéficier de cette prise en charge optimale et de se voir possiblement proposer l’inclusion dans des essais cliniques innovants.
Références
1. de Pinieux G, Karanian M, Le Loarer F, Le Guellec S, Chabaud S, Terrier P, et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. PLoS One 2021;16(2):e0246958.
2. Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers 2021;7(1):22.
3. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida Tn Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279 (5350):577-80.
4. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299(5607):708-10.
5. Casali PG, Blay JY, Abecassis N, Bajpai J, Bauer S, Biagini R, et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2022;33(1):20-33.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol 2008;39(10):1411-9.
7. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: A randomized trial. JAMA 2012;307(12):1265-72.
8. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347(7):472-80.
9. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006;368(9544):1329-38.
10. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):295-302.
11. Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2020;21(7):923-34.
12. Heinrich MC, Jones RL, von Mehren M, Schöffski, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol 2020;21(7):935-46.
13. Choi H. Response evaluation of gastrointestinal stromal tumors. Oncologist 2008;13(Suppl 2):4-7.
2. Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers 2021;7(1):22.
3. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida Tn Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279 (5350):577-80.
4. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299(5607):708-10.
5. Casali PG, Blay JY, Abecassis N, Bajpai J, Bauer S, Biagini R, et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2022;33(1):20-33.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol 2008;39(10):1411-9.
7. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: A randomized trial. JAMA 2012;307(12):1265-72.
8. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347(7):472-80.
9. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006;368(9544):1329-38.
10. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):295-302.
11. Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2020;21(7):923-34.
12. Heinrich MC, Jones RL, von Mehren M, Schöffski, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol 2020;21(7):935-46.
13. Choi H. Response evaluation of gastrointestinal stromal tumors. Oncologist 2008;13(Suppl 2):4-7.
Dans cet article

Résumé
Les tumeurs stromales gastro-intestinales (GIST) représentent le sous-type de sarcomes le plus fréquent. Plus de 80 % des GIST sont caractérisées par des mutations activatrices des gènes KIT ou PDGFRA, mais des sous-types moléculaires plus rares existent. Au stade localisé, les GIST sont des maladies curables par exérèse chirurgicale. Le traitement adjuvant par imatinib est un standard thérapeutique dans les GIST associées à un haut risque de récidive et présentant des mutations sensibles au traitement. Au stade métastatique, le développement des inhibiteurs de tyrosine kinase ciblant KIT et PDGFRA a bouleversé la prise en charge et le pronostic des patients, en augmentant la survie globale : de moins de dix-huit mois il y a une vingtaine d’années à plus de soixante-dix mois aujourd’hui. Comme pour les autres sous-types histologiques, la prise en charge diagnostique et thérapeutique des GIST doit être réalisée dans des centres experts pour la prise en charge des sarcomes.