Expliquer les bases du conseil génétique, les possibilités de diagnostic prénatal (modalités et options de prise en charge dans le cadre d'une maladie d'une particulière gravité).
Expliquer la prescription des tests génétiques : organisation et aspects réglementaires (voir item 8).
Expliquer les problèmes liés à la maladie et les retentissements de l'arrivée d'un enfant souffrant de maladie génétique sur le couple et la famille.
Diagnostiquer la trisomie 21, en connaître l'évolution naturelle et les principales complications.
Introduction
Un diagnostic génétique permet une prise en charge à multiples « dimensions ». S’approprier ces notions est essentiel pour la prise en charge des patients :
- une dimension diagnostique de précision, permettant de déterminer la cause de la maladie et d’y mettre un nom ;
- une dimension pronostique, un diagnostic permettant une prise en charge préventive et d’accompagnement personnalisé des patients, grâce à la trajectoire de maladie des patients ;
- une dimension d’aide à la prescription thérapeutique (thérapies ciblées, pharmacogénomique) ;
- une dimension familiale avec parfois obligation d’informer les membres de sa famille pour qu’ils puissent bénéficier d’un conseil génétique adapté (décret parentèle) ;
- une dimension liée à la reproduction, avec la réalisation d’un diagnostic prénatal (DPN) ou pré-implantatoire (DPI), la possibilité de réaliser une interruption médicale de grossesse (IMG). Enfin, le patient peut avoir connaissance de son statut hétérozygote de maladies génétiques récessives telles que la mucoviscidose. Cette découverte a une implication pour sa descendance (dépistage préconceptionnel).
Déroulement de la consultation pour prescription et rendu des résultats
- d’apporter des explications sur la maladie génétique du patient ;
- d’évaluer un risque de survenue ou de récurrence de cette maladie génétique pour une future grossesse au sein de la famille (pour le patient et ses apparentés) ;
- de proposer des solutions de prévention de cette récurrence ;
- d’apporter les informations sur la prise en charge personnalisée avec les différents spécialistes d’un centre expert de la maladie génétique ;
- d’informer sur l’existence d’associations de patients de la maladie identifiée.
Enquête génétique
Prescription d’analyses génétiques
Rendu de résultats : explication de la maladie, hérédité et prise en charge
Une information à la parentèle est à réaliser si celle-ci peut apporter un bénéfice à la famille.
Actuellement, il n’existe dans la plupart des maladies génétiques aucune solution thérapeutique curative. La prise en charge coordonnée des handicaps possibles du patient est néanmoins essentielle. Un diagnostic génétique facilite les démarches administratives de prise en charge auprès de la maison départementale des personnes handicapées (MDPH) [aides financières – allocation d’éducation de l’enfant handicapé, AEEH –, aides logistiques, aménagement et orientation scolaire...]. Un suivi par une structure adaptée scolaire (classe spécialisée intégrée [CLIS], section d’enseignement général et professionnel adapté [SEGPA]...) et médicale (centre de référence maladies rares, centre d’action médico-sociale précoce [CAMSP], service d’éducation spécialisée et de soins à domicile [SESSAD], institut médico-éducatif [IME]) est primordial pour un accompagnement adapté et une prise en charge des symptômes.
Un diagnostic de précision permet également d’éviter des examens et des traitements inutiles (informations apportées lors de la demande de prise en charge en affection de longue durée hors liste de la Caisse primaire d’assurance maladie [CPAM]). L’ensemble de cette prise en charge est souvent éprouvante pour les familles, nécessitant du temps et un aménagement familial ainsi que professionnel des parents et proches. Une prise en charge psychologique est souhaitable.
Conseil génétique : information au couple du risque de récurrence, perception et aide à la prise de décision
La perception de ce risque est très différente selon les couples (perception de la gravité de la maladie, expérience de la maladie au sein de la famille…).
En cas de souhait de nouvelle grossesse et de l’identification d’un risque, le praticien doit informer le couple de l’ensemble des solutions de prévention dans le cadre réglementaire et des possibilités techniques :
- un diagnostic prénatal avec la compréhension d’une possible interruption médicale de grossesse (IMG) ;
- un diagnostic pré-implantatoire (DPI) ;
- le recours à un don de gamètes ou à une adoption ;
Prescription génétique
Recueil de l’attestation de consultation et du consentement par écrit
Partage de l’information au sein de la famille
Cette disposition s’applique également pour les enfants de cette personne à l’issue d’un don de gamètes via le centre d’assistance médicale à la procréation.
Rendu du résultat et secret professionnel
Cas particulier du diagnostic présymptomatique
Diagnostic anténatal
Diagnostic prénatal
À l’issue de cette exploration étiologique, si le couple a « une forte probabilité de donner naissance à un enfant atteint d’une maladie génétique d’une particulière gravité reconnue comme incurable au moment du diagnostic », il est possible de proposer à titre exceptionnel une IMG. Sa réalisation et sa décision sont faites au sein de centre pluridisciplinaire de diagnostic prénatal (CPDPN) agréé par l’Agence de la biomédecine. La demande d’IMG est formulée par le couple. La décision est prise de façon collégiale par les médecins du CPDPN.
Diagnostic pré-implantatoire
Trisomie 21
Physiopathologie et diagnostic biologique
Le diagnostic est confirmé par le caryotype sur culture cellulaire (seul examen permettant de déterminer la mécanique chromosomique).
Signes cliniques et principales complications (tableau 1)
Les personnes porteuses d’une trisomie 21 ont une espérance de vie supérieure à 60 ans, limitée par une démence précoce de type Alzheimer (30 %-50 %) par triplication du gène APP. Les principales complications sont liées à l’hypotonie, une susceptibilité aux infections, un contexte auto-immun et aux malformations congénitales.
Prise en charge
- prise en charge médicale avec surveillance des complications éventuelles (
v. supra ) ;
- prise en charge éducative et paramédicale : kinésithérapie (hypotonie et hyperlaxité), psychomotricité (adaptation sociale), orthophonie (hypotonie et dentition) et psychologique (pour la famille et l’enfant) ;
- prise en charge éducative : accompagnement en milieu ordinaire ou adapté, aide au développement des relations sociales, accompagnement autour de la vie affective et sexuelle.
Conseil génétique
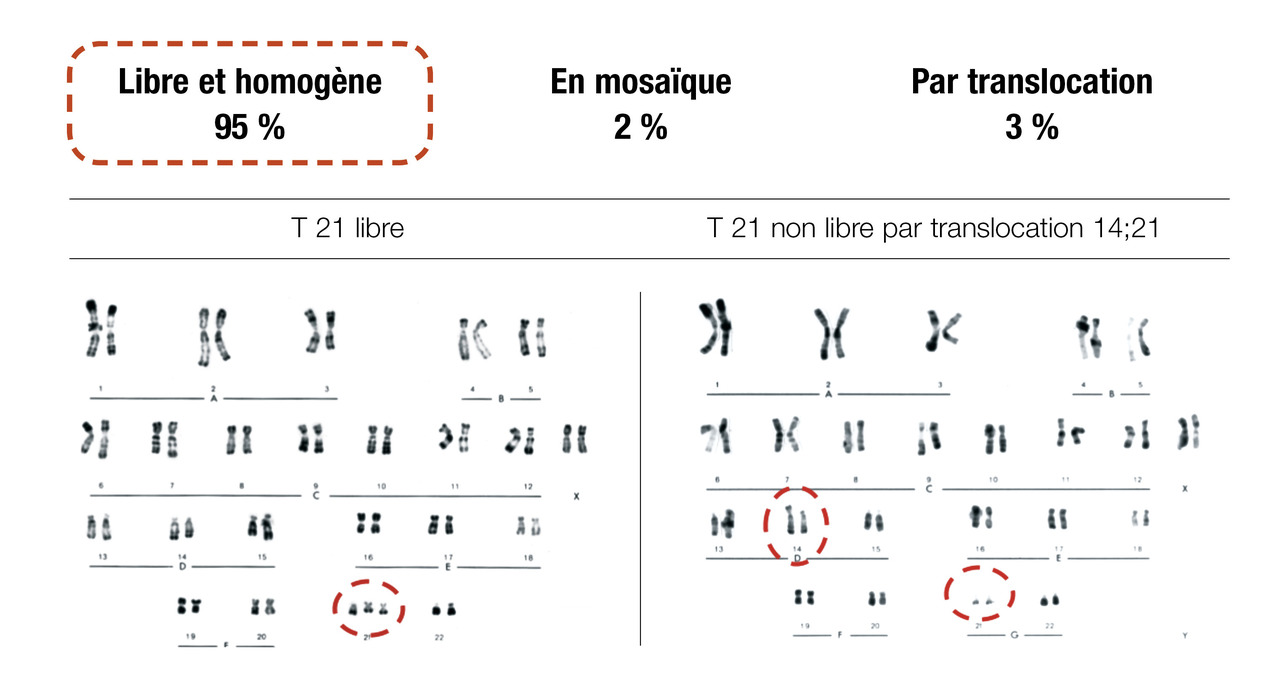
Trisomie 21 libre et homogène (95 % des cas) : il s’agit une anomalie de ségrégation des chromosomes lors de la formation des gamètes (méiose). Il s’agit d’un « accident génétique » de novo, le risque de récurrence est faible (1 % avant 40 ans) et donc l’analyse génétique des parents n’est pas préconisée. Le seul facteur de risque connu est l’âge maternel (1/1 000 à 30 ans, 1/100 à 40 ans).
Trisomie 21 libre en mosaïque (2 %) : seul un contingent de cellules porte la trisomie 21. L’autre contingent est euploïde. Le risque de transmission est difficile à déterminer.
Trisomie 21 non libre (3 % des cas) : il peut s’agir également d’un « accident génétique » de novo (risque de récurrence faible) ou alors par transmission d’un parent porteur d’une anomalie chromosomique équilibrée (translocation réciproque ou translocation robertsonienne, avec un risque de récurrence plus élevé de 5 % [pour l’homme] à 15 % [pour la femme]. En cas de translocation 21-21, le risque est de 100 %). Une exploration des parents est alors indiquée.
Patient porteur d’une trisomie 21 : risque de 30 % chez les femmes porteuses (pertes embryonnaires précoces). Pas de transmission décrite pour les hommes porteurs.
Dépistage prénatal
- entre 11 SA et 13+6 SA : bêta-human chorionic gonatropin (bêta-hCG) et pregnancy associated plasma protein A (PAPP-A) [dépistage combiné, recommandé] ;
- entre 14 SA et 17+6 SA : bêta-HCG et alpha-fœtoprotéine (a-FP) [dépistage séquentiel, si combiné non réalisable].
- si le risque du dépistage intégré est supérieur à 1/50, de réaliser un caryotype fœtal par amniocentèse afin d’affirmer ou d’infirmer le diagnostic de trisomie 21. Le taux de fausse couche d’une amniocentèse est estimé à moins de 1 % ;
- si le risque est entre 1/51 et 1/1 000, de réaliser un dépistage non invasif sur sang maternel, basé sur le dosage chromosomique par étude de l’ADN fœtal circulant (technique de séquençage haut débit). Si cette méthode (fiable à plus de 99 %) montre un excès de séquence du chromosome 21, un caryotype fœtal par amniocentèse doit être réalisé afin d’affirmer le diagnostic de trisomie 21.
Mucoviscidose
Physiopathologie et diagnostic
Les variants pathogènes (ou mutations, terme ancien) sont répertoriés en 6 classes selon le mécanisme d’atteinte du gène CFTR. Il existe une tentative de corrélation pronostique à partir de ces classes (classe 1, 2, 3 = variants pathogènes, dite sévère ; classe 4, 5 = variants pathogènes, peu sévère). Les corrélations génotype/phénotype doivent néanmoins être maniées avec prudence pour le pronostic et le DPN, car au plan individuel il existe des patients « peu sévères » avec des variants pathogènes sévères et vice-versa. Avoir un diagnostic précis permet aussi de proposer des thérapies spécifiques selon la classe mutationnelle.
La mutation la plus fréquente (70 % des patients) dans ce gène est une délétion de 3 paires de base (respectant le cadre de lecture) entraînant la perte d’une phénylalanine en position 508 (F508del, classe 2). Près de 2 000 variants pathogènes sont répertoriés, en majorité des variants pathogènes faux-sens (ou de substitution) dont la fréquence dépend des populations (variants différents chez les Bretons, chez les Juifs ashkénazes…).
Un dépistage néonatal systématique de la mucoviscidose est réalisé. Il consiste au 3e jour de vie au dosage de la trypsine immuno-réactive, couplée à la recherche des variants pathogènes les plus fréquents (nombre dépend des kits) si le dosage est élevé (seuil à 60 mEq, soit 1/200 enfants avec test génétique). En cas d’identification de 2 variants pathogènes (1 homozygote ou 2 hétérozygotes), chacun hérité des 2 parents, le diagnostic de mucoviscidose est posé.
En cas d’identification d’une seule mutation, un test de la sueur est proposé (positif si > 60 mEq/L, négatif si < 30 mEq/L). S’il est positif, une étude complète du gène par séquençage haut débit est réalisée.
Signes cliniques (tableau 2)
Le principal facteur pronostique est le début de la colonisation par Pseudomonas aeruginosa.
Les patients atteints de variants pathogènes sévères ont une espérance de vie d’environ 30 ans et de 50 ans en cas de variants pathogènes peu sévères.
CFTR-related disorders (CFTR-RD)
- stérilité masculine par agénésie bilatérale des canaux déférents (ABCD) ;
- pancréatite chronique ;
- dilatation des bronches isolée à un âge tardif.
Prise en charge
- prise en charge multidisciplinaire avec éducation thérapeutique (signes d’alerte, règles d’hygiène afin de limiter une colonisation bactérienne) ;
- prise en charge respiratoire avec kinésithérapie, aérosolthérapie, activité physique, antibiothérapie inhalée. En cas de pneumopathie, une antibiothérapie basée sur le type de germe est entreprise ;
- prise en charge nutritionnelle, veille aux apports hydrosodés (notamment si sport), alimentation adaptée aux carences, extraits pancréatiques.
Thérapie ciblée
- ivacaftor (Kalydeco) : mutation G551D (classe 3) et 7 autres variants ;
- ivacaftor + lumacaftor (Orkambi) : mutation F508del homozygote (50 % de la population européenne).
Conseil génétique
- 2 parents hétérozygotes connus, porteurs asymptomatiques : risque de récurrence à 25 % ;
- aucun statut connu : le risque de la population générale est de 1/4 000 (1/33* 1/33* 1/4) ;
- au vu de la fréquence des hétérozygotes dans la population générale (1/33), il est fortement recommandé de tester le conjoint d’une personne porteuse. Pour plus d’informations sur les calculs de risque, v. référentiel.
Diagnostic prénatal
Si chaque parent du couple est porteur d’une mutation pathogène, un diagnostic prénatal par biopsie de trophoblaste dès 12 SA ou par amniocentèse dès 15 SA est possible. Si le fœtus est atteint, la décision d’une IMG peut être accompagnée au sein d’un CPDPN.
Une démarche de diagnostic préimplantatoire (DPI) est également possible.
Syndrome de l’X fragile
Physiopathologie et diagnostic biologique
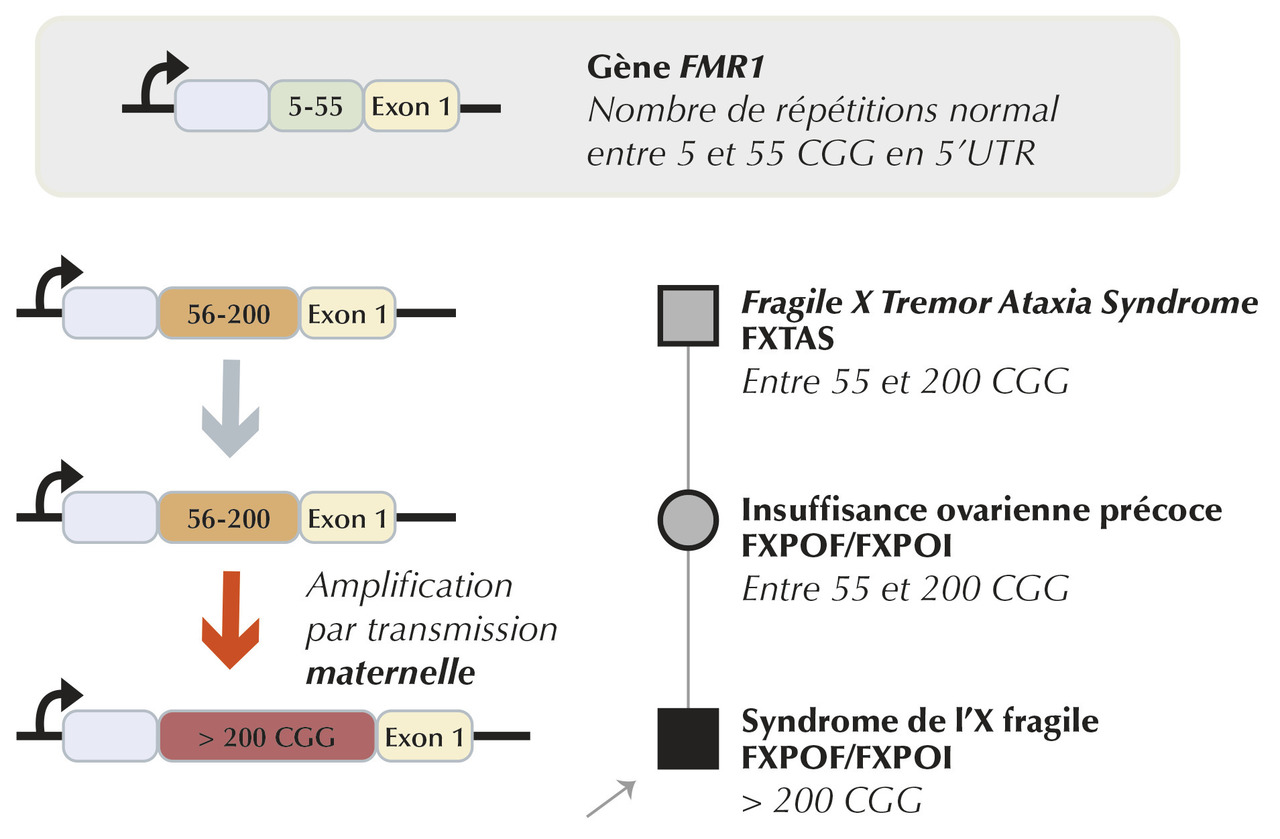
Dans sa forme courante, cette altération est liée à une expansion anormale de répétitions du trinucléotide CGG, situé dans la région 5’UTR du gène FMR1.
Très rarement, il peut y avoir une délétion du gène ou des variants pathogènes ponctuelle.
Le syndrome de l’X fragile doit son nom à une ancienne technique cytogénétique utilisée pour son diagnostic.
Il existe 2 types de variations pathologiques familiales :
- la norme est un nombre de triplet < 50 CGG ;
- les prémutations contenant entre 55 et 200 trinucléotides CGG. Une accentuation de cette expansion avec le passage à une mutation complète n’est possible que lors de transmissions maternelles. Ce risque est d’autant plus important que le nombre de répétitions de la prémutation est élevé ;
- les mutations complètes sont associées à une hyperméthylation de la région promotrice entraînant l’arrêt de la transcription du gène FMR1 au-delà de 200 trinucléotides CGG.
Pour le diagnostic de porteur d’une prémutation, il est nécessaire de suivre une procédure de test présymptomatique (v. supra) dû au risque de FXTAS et FXPOF (v. infra).
Le test génétique de mineur asymptomatique n’est pas indiqué.
Signes cliniques du syndrome de l’X fragile
Les signes cliniques chez les garçons sont rapportés dans le
Chez les filles et femmes conductrices avec mutation complète : 30 à 40 % des femmes conductrices n’ont pas de déficit intellectuel ; 60 % ont des difficultés cognitives de sévérité très variable. Cette variabilité de la déficience intellectuelle est probablement expliquée par le taux d’inactivation du chromosome X avec l’anomalie du gène FMR1. Des troubles des relations et des troubles anxieux peuvent être associés.
Signes cliniques des patients porteurs d’une prémutation
FXTAS (fragile X-associated tremor ataxia syndrome)
L’incidence est de 16 % des femmes et 40 % des hommes porteurs.Facteur de risque : le risque est augmenté en cas de prémutation au-delà de 70 CGG.
Il existe une association variable au-delà de 60 ans à un tremblement (d’action ou repos), une ataxie cérébelleuse, un syndrome parkinsonien, une neuropathie sensitive, des troubles cognitifs et des troubles psychiatriques.
FXPOI ou FXPOF (fragile X-associated primary ovarian insufficiency or failure)
L’incidence est de 20 % des femmes porteuses d’une prémutation.Facteur de risque : augmente entre 70 et 100 CGG puis diminue au-delà.
Les porteuses peuvent présenter une insuffisance ovarienne précoce avant 40 ans pouvant entraîner une ménopause précoce et des difficultés de procréation.
Son diagnostic précoce permet une exploration de la réserve ovarienne, un suivi hormonal et éventuellement une cryoconservation d’ovocytes en vue d’un DPI.
Conseil génétique et diagnostic prénatal
Hérédité : homme porteur d’une prémutation : le risque de transmission est de 100 % pour ses filles et 0 % pour ses garçons ; femme porteuse d’une prémutation : le risque de transmission est de 50 % pour ses garçons et ses filles de sa prémutation ou d’une mutation complète ; femme porteuse d’une mutation complète : le risque de transmission est de 50 % pour ses garçons et ses filles.
Diagnostic prénatal : un DPN ou un DPI sont possibles uniquement pour une femme porteuse d’une prémutation ou d’une mutation complète, à risque d’avoir un garçon atteint du syndrome de l’X fragile. Une IMG ou une sélection d’embryons n’est pas proposée en cas de prémutations. •
Problèmes posés par les maladies génétiques, à propos : – d'une maladie chromosomique : la trisomie 21 – d'une maladie génique : la mucoviscidose – d'une maladie d'instabilité : le syndrome de l'X fragile
Le prescripteur a pour obligation légale de préciser avant le prélèvement : le nom du gène ou du groupe de maladie recherché, la prise en charge multidisciplinaire, le mode d’hérédité de la maladie pour identifier les autres membres potentiellement atteints (décret parentèle) et la réalisation ou non du don de gamètes.
La déficience intellectuelle (QI < 70) atteint 1 personne sur 40 en population générale, majoritairement due à des causes génétiques. La trisomie 21 et le syndrome de l’X fragile en sont les causes les plus fréquentes.
Le dépistage de la trisomie 21 a radicalement changé grâce au dépistage prénatal non invasif (DPNI), proposé en cas de risque combiné entre 1/51 et 1/1 000. Les complications mettant en jeu le pronostic vital sont : les malformations cardiaques congénitales, l’atrésie digestive, la maladie de Hirschsprung, la leucémie (LAM7). Tous les adultes développent une maladie d’Alzheimer par triplication du gène APP.
La mucoviscidose est la maladie génétique récessive grave la plus fréquente avec le plus fort taux de porteurs hétérozygotes en population générale (1/33).
Le syndrome de l’X fragile est réservé aux hommes et les femmes avec mutation complète de répétitions CGG. Les femmes porteuses d’une prémutation ont un risque de développer une FXPOF (20 %). Les hommes et les femmes porteurs d’une prémutation ont un risque de développer un FXTAS (16 % femmes, 40 % hommes).
Martin Krahn et Damien Sanlaville. Génétique médicale : enseignement thématique, Elsevier-Masson, 2016.
Références de la Haute Autorité de santé :
– PNDS mucoviscidose 2017 ;
– Recommandations en santé publique. Place des tests sur ADN libre circulant dans le sang maternel dans le dépistage de la trisomie 21 fœtale, 2017.
 Encadrés
Encadrés