D’après :
Alexandre M. Pemphigoïde bulleuse. Rev Prat Med Gen 2017 ;31(989) :731 - 3.
Hebert V, Joly P. La pemphigoïde bulleuse, une dermatose du sujet âgé. Rev Prat 2017 ;67(10) :1080 - 3.
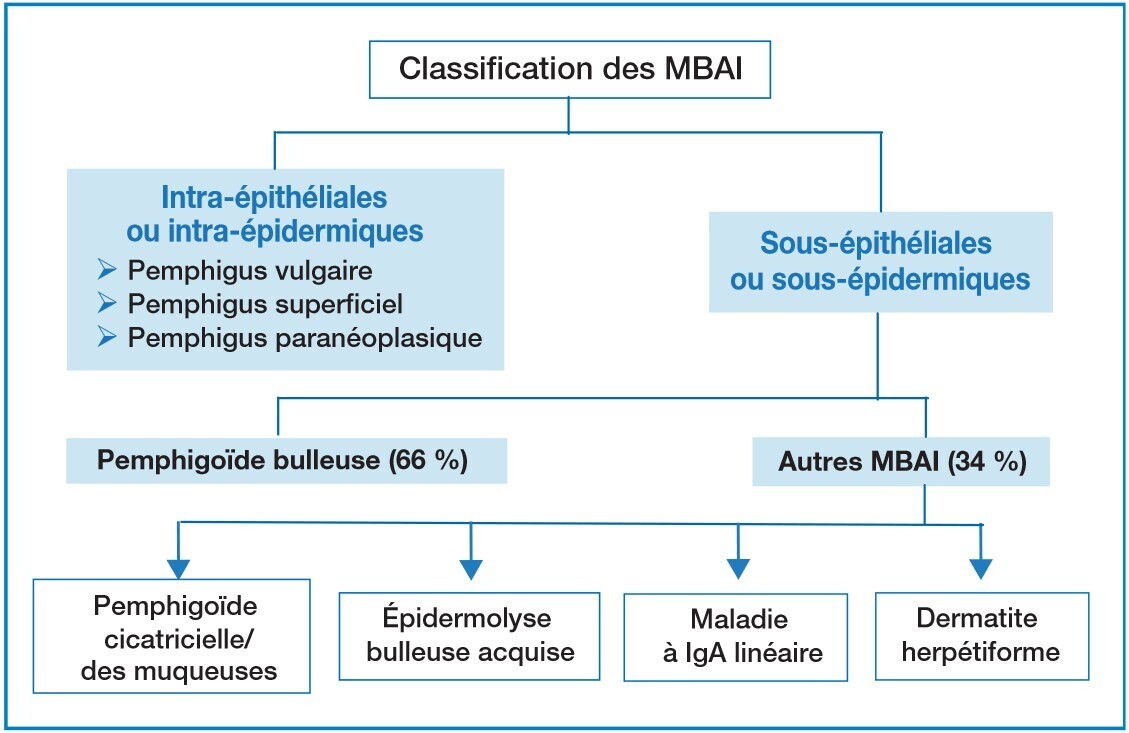
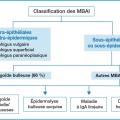
Les maladies bulleuses auto-immunes (MBAI) sont un groupe hétérogène de maladies à la fois très diverses, peu fréquentes et de pronostic variable, liées aux lésions de différents constituants de la peau (fig. 1). La plus fréquente est la pemphigoïde bulleuse (PB) : c’est une pathologie du sujet âgé se révélant en moyenne entre 75 et 81 ans. L’association à des pathologies neurodégénératives est fréquente : démence, AVC, maladie de Parkinson.
La pemphigoïde bulleuse est liée à la production d’autoanticorps IgG1 et/ou IgG4, dirigés contre l’antigène BPAG1 (ou BP 230) situé dans les hémidesmosomes, et contre la glycoprotéine transmembranaire BPAG2 (ou BP 180) au niveau de son épitope NC 16A (localisé au contact de la membrane des kératinocytes dans la lamina lucida). L’altération de ces deux constituants de la jonction dermo-épidermique (JDE), provoquée par la fixation des auto-anticorps, entraîne la formation d’une bulle sous-épidermique. Les lésions résultent également du recrutement et de l’activation de cellules pro-inflammatoires – polynucléaires éosinophiles et mastocytes – qui libèrent des substances capables de dégrader les systèmes de jonction. En outre, les IgE jouent, au moins chez certains patients, un rôle pathogène.
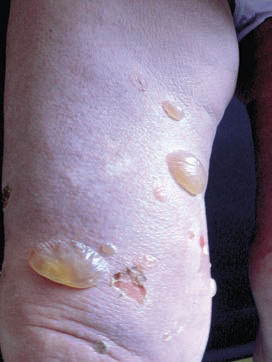
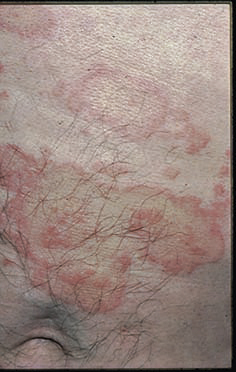
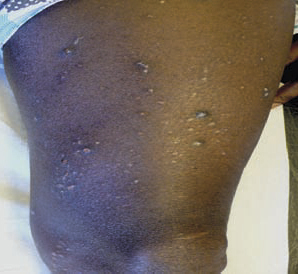
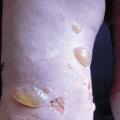
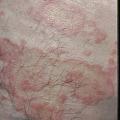
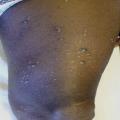
Le diagnostic de la forme typique (fig. 2) est aisé : survenue chez le sujet âgé (> 70 ans) de bulles spontanées de grande taille, tendues, sur une base érythémateuse, associées à des lésions urticariennes ou eczématiformes, siégeant de façon bilatérale et globalement symétrique sur les faces de flexion des membres, les plis inguinaux et axillaires, la partie basse de l’abdomen. L’extrémité céphalique est habituellement épargnée. Leur nombre est très variable : de moins de 10 (formes paucibulleuses) à plus de 300 (multibulleuses) ; elles laissent place, une fois rompues, à des érosions. Il faut les comptabiliser quotidiennement. Le prurit, volontiers intense, est quasi constant et précède souvent l’éruption. Les lésions guérissent sans laisser de cicatrice. L’atteinte muqueuse est rare (8 % des cas) et doit faire évoquer un diagnostic différentiel (encadré 1). L’hémogramme montre habituellement une hyperéosinophilie. Des formes atypiques sont décrites : atteintes chroniques aspécifiques eczématiformes ou urticariennes fixes pré- ou post-bulleuses : (fig. 3), tableau de prurigo chez un sujet âgé (fig. 4), prurit sine materia.
Diagnostic : quels examens ?
L’histologie (biopsie sous anesthésie locale d’une bulle intacte ou récente ou à défaut de la bordure d’une érosion) confirme le diagnostic de maladie bulleuse de la jonction en montrant une bulle sous-épidermique contenant des polynucléaires neutrophiles et/ou surtout éosinophiles. L’immunofluorescence directe (IFD) sur biopsie de la peau péribulleuse met en évidence des dépôts immuns linéaires d’IgG et/ou de complément (C3) à la JDE/JCE. L’immunomicroscopie électronique (IME), disponible uniquement dans les centres de référence, et les examens sérologiques ne sont ni nécessaires ni suffisants pour porter le diagnostic. Ainsi, devant une MBAI sous-épidermique prouvée en IFD, le diagnostic de pemphigoïde bulleuse est clinique et repose sur les critères (encadré 2).
Quelle prise en charge ?
L’âge élevé des malades et leurs comorbidités (neurologiques, cardiovasculaires, néoplasiques, métaboliques, respiratoires…) rendent la prise en charge particulièrement délicate. Il est essentiel de confirmer le diagnostic, notamment par IFD, avant la mise en place d’une corticothérapie locale ou générale, afin de ne pas risquer de négativer les résultats.
Le contrôle de l’éruption bulleuse (comptage quotidien) est l’objectif essentiel en essayant de minimiser autant que possible les effets indésirables graves des traitements.
Traitement de choix : un dermocorticoïde (DC) à forte dose administré pendant plusieurs mois. En phase d’attaque, on préfère le propionate de clobétasol crème (Dermoval, 20 à 40 g/j soit 2 à 4 tubes/j, selon la corpulence du patient et le nombre de bulles), appliqué sur tout le corps y compris les érosions en n’épargnant que le visage, les organes génitaux et les plis. Il a montré une efficacité similaire à la corticothérapie générale à 1 mg/kg/j, avec une meilleure tolérance (diminution des complications iatrogènes et de la mortalité).
Aux DC doivent être associés :
– des soins locaux : toilette antiseptique si surinfection, perçage et assèchement des bulles avec du nitrate d’argent en solution aqueuse, pansements gras ou siliconés sur les érosions (pas de pansements adhésifs en raison de la grande fragilité cutanée).
– des mesures générales : prévention des complications de décubitus, lutte contre la dénutrition, compensation des pertes hydroélectrolytiques, éradication des foyers infectieux, mise à jour vaccinale, surveillance et dépistage des effets secondaires éventuels de la corticothérapie au long cours (diabète, HTA, insuffisance surrénale…).
En cas de résistance ou de dépendance aux DC, un traitement immunosuppresseur peut être proposé ou, en cas de contre-indication, un immunomodulateur.
Le pronostic est réservé, avec une mortalité d’environ 20 - 40 % à 1 an. Il dépend essentiellement du terrain : le mauvais état général, le grand âge et les comorbidités neurologiques sont les principaux facteurs de risque de mortalité. La prise en charge des comorbidités et la surveillance étroite des effets indésirables de la corticothérapie sont donc primordiales.
Qu’en retenir ?
Les formes pré- et post-bulleuses pouvant être trompeuses, il faut y penser devant un prurit persistant ou des lésions urticariennes/eczématiformes chez un sujet âgé.
Son diagnostic repose sur l’IFD associée à des critères cliniques.
C’est une maladie chronique nécessitant une prise en charge et un suivi sur plusieurs mois. Le traitement est efficace mais les rechutes sont fréquentes.
Appliquer correctement la corticothérapie locale est crucial.
Il faut, dans la mesure du possible, arrêter les médicaments inducteurs (diurétiques en particulier).
Diagnostics différentiels
- MBAI intra-épidermiques : pemphigus (herpétiforme notamment).
- Autres MBAI sous-épidermiques : pemphigoïde cicatricielle/pemphigoïde des muqueuses, épidermolyse bulleuse acquise, maladie à IgA linéaire…
- Pathologies bulleuses non auto-immunes (IFD négative) : eczéma dans une forme bulleuse, impétigo bulleux, toxidermies bulleuses, bulles hydrostatiques ou de pression, bulleuse du diabétique…
Critères diagnostiques de pemphigoïde bulleuse
Devant une MBAI sous-épidermique prouvée en IFD, la présence de 3 des 4 critères suivants fait poser le diagnostic de PB avec une sensibilité de 90 % et une spécificité de 83 % :
- Âge supérieur à 70 ans ;
- Pas de cicatrice atrophique ;
- Pas d’atteinte muqueuse ;
- Pas de prédominance des lésions à la tête et au cou.
 Encadrés
Encadrés